

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cellular senescence is defined as a state in which normal somatic cells lose their ability to divide, due to either prolonged cell division in culture or exposure to subcytotoxic stressors, thereby entering irreversible growth arrest [1]. In addition to loss of replicative capacity, senescent cells display alterations in morphology, mass, and the dynamics of subcellular organelles [2-4]. For example, increases in cellular mass have been documented as a typical feature of senescence and are known to be associated with enlarged cell morphology. However, it is unclear which molecular events have critical roles in these features. An increase in the mass of any specific cellular compartment has been suggested as a potential cause of senescence. For example, lysosomes have long been recognized to increase in both senescent and aged cells [5,6]. Increased mitochondrial mass has also been reported in stress-induced senescence systems [7,8]. These phenomena were thought to be the result of an accumulation of damaged organelles, because senescent lysosomes are known to be nonfunctional [9]. However, the accumulation of indigestible organelles is not sufficient to explain the enlarged morphology observed in senescent cells. We suggested a more plausible explanation based on the results of our recent study, which found that enhanced lipogenesis in response to expression of sterol regulatory element binding protein 1 (SREBP1, also known as SREBF1) is involved in the increase in the organellar mass observed in senescent cells [10].
SREBP1 is a master transcription factor that regulates the expression of several lipogenic enzymes, including ATP citrate lyase (ACL), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FAS) [11]. During lipid synthesis, ACL converts cytosolic citrate into acetyl-CoA and oxaloacetate, the essential metabolites for lipid biosynthesis, and ACC carboxylates acetyl-CoA to produce malonyl-CoA. FAS then condenses acetyl-CoA and malonyl-CoA to generate long-chain fatty acids. Thus, activation of SREBP1 effectively enhances overall lipid biosynthesis, including fatty acid synthesis and the subsequent synthesis of triglycerides and membrane phospholipids. Enhanced lipogenesis provides the essential membrane components for the formation of subcellular organelles during cell growth [12,13]. SREBP1 is initially localized to the rough endoplasmic reticulum (ER) membrane. Upon activation, it is cleaved to liberate the N-terminal fragment, which contains a basic helix-loop-helix leucine-zipper domain. The cleaved active fragment, called mature-form SREBP1 (mSREBP1), enters the nucleus, where it binds to specific sterol response elements within the promoter regions of the lipogenic genes and activates their transcription [11,14]. Thus, SREBP1 is an excellent target molecule for studies of cellular lipogenesis regulation.
Glycogen synthase kinase 3 (GSK3) is a serine/threonine kinase that is involved in the control of a wide range of cellular metabolic processes, including insulin sensitivity, glucose metabolism, and cell survival, via regulation of energy homeostasis [15,16]. In addition, it is a multifunctional kinase that performs a regulatory role in diverse cellular functions, such as embryonic development, cellular protein synthesis, mitosis, and survival [16-18]. GSK3 comprises two isoforms, GSK3α and GSK3β, which are negatively regulated by phosphorylation (inactivation) at Ser21 (GSK3α) or Ser9 (GSK3β) by several protein kinases, such as AKT and p90 ribosomal S6 kinase [19]. Although the involvement of GSK3 inhibition in Wnt signaling implies that it has a role in cell proliferation and cancer development [20,21], the death of GSK3β-null mice during embryogenesis as a result of liver degeneration caused by widespread hepatocyte apoptosis also suggests a contrasting role for GSK3, including participation in senescence-associated growth suppression [22]. Recently, we reported that GSK3-mediated glycogenesis is involved in cell senescence [23], which directly supports the importance of GSK3 modulation in cell senescence. Based on these reports, we can place GSK3 downstream of the key insulin like growth factor-1 receptor (IGF-1R)/phosphoinositide 3-kinase (PI3K)/AKT signaling pathway, which is known to regulate cellular and organismal lifespans [24-28].
We previously reported that levels of the mature form of SREBP1 are commonly increased in Chang cells during cellular senescence induced by several stimuli, including deferoxamine (DFO) and hydrogen peroxide (H2O2). Under these conditions, SREBP1-mediated lipogenesis is critically involved in cellular senescence [10]. However, it was unclear which signaling molecules control the SREBP1-mediated senescence that accompanies enhanced lipogenesis. Recently, GSK3 was shown to modulate SREBP1 activity through phosphorylation-mediated ubiquitination [29]; this implies that GSK3 may play a key role as an upstream regulator in SREBP1-mediated senescence. To address this, we first developed a model for stress-induced senescence by exposing Chang cells, which are immortalized human liver cells, to subcytotoxic concentrations (200 µM) of DFO and H2O2, as described previously [7,30]. By employing these senescence systems, we demonstrated that GSK3 is one of the major regulators of SREBP1-mediated lipogenesis, and its inactivation contributes to the increase in the organellar mass that is a typical characteristic of senescence. Our results suggest a novel molecular background for aging and aging-related pathophysiology.
METHODS
Cell culture, cell growth rate and generation of stress-induced senescence
Chang cells were purchased from the American Type Culture Collection (Rockville, MD, USA) and were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (GIBCO, Grand Island, NY, USA) and antibiotics at 37℃ in a humidified incubator with 5% CO2. Chang cells, an immortalized normal human hepatocyte line, were chosen to understand metabolic backgrounds related to stress-induced senescence.
To evaluate cellular growth rate, cells were harvested by trypsinization and counted with a hemocytometer after staining with 0.4% (w/v) trypan blue (GIBCO) to exclude dead cells. The number of dead cells observed in the progress of the two different stress-induced senescence systems was not significant. The total cell number of trypan blue-negative live cells was determined upon completion of the experiments.
Estimation and visualization of subcellular organellar mass
To estimate cellular organellar mass, cells were stained with organelle-specific fluorescence dyes according to the manufacturer's instruction. Briefly, proper concentrations and incubation periods for each organelle were first determined. Mitotracker Red CMXRos (100 nM, M7512, Molecular Probes, Eugene, OR, USA), Lyso Tracker Green DND-26 (1 µM, L7526, Molecular Probes), and ER Tracker Blue-White DPX (1 µM, E-12353, Molecular Probes) were applied for 30 minutes at 37℃. To stain Golgi body, cells were incubated for 30 minutes at 4℃ with 5 µM boron-dipyrromethene C5-ceramide (D3521, Molecular Probes) after preconjugating the dye with bovine serum albumin and were further incubated in fresh medium at 37℃ for 30 minutes. After staining cells with the dyes, cells were harvested in phosphate-buffered saline (PBS), and their fluorescence intensities were analyzed by flow cytometry (FACSCanto II, Becton Dickinson Corp., San Jose, CA, USA). Subcellular organellar masses are represented as mean value of arbitrary fluorescence units of 10,000 cells.
To confirm specific staining of subcellular organelles with the dyes, fluorescence images of the stained cells were visualized by Axiovert 200 M with a cooled AxioCam HRm operated by the Axiovision 4.8 software (Carl Zeiss AG, Gottingen, Germany).
Determination of intracellular reactive oxygen species, cell size, and cell granularity
To estimate intracellular reactive oxygen species level, cells were stained with 10 µM 2',7'-dichlorodihydrofluorescein diacetate (Molecular Probes) for 15 minutes at 37℃ before performing flow cytometric analysis with FACSCanto II (Becton Dickinson Corp.). Cell size and granularity were evaluated by analyzing forward scatter and side scatter of the stained cells, respectively, as previously described [10].
Thin layer chromatographic analysis of cellular lipids
To analyze cellular lipid profiles, cellular lipids were extracted as described by Folch et al. [31] with slight modification. Briefly, the indicated numbers of cells were harvested into glass tubes and mixed with 1 mL of chloroform:methanol (2:1). After vortexing, distilled water (500 µL) was added into the tubes and centrifuged at 1,500 rpm for 15 minutes. The lower chloroform phase was collected and backwashed by adding methanol (300 µL) and distilled water (300 µL). After removing the top layer, chloroform phase were concentrated by evaporating chloroform under N2 gas and were loaded onto thin layer chromatography (TLC) plate (no. 1.05721.0001, 20×20, silica-coated, Merk, Darmstadt, Germany).
To separate the cellular lipids, we used Kupke and Zeugner's method [32] with a slight modification. Briefly, the chromatogram was first developed in a saturated chamber with solvent I (chloroform:methanol:H2O, 65:30:5) to allow separation of polar lipids. The solvent front was allowed to migrate approximately 10 cm from the bottom of the plate. After evaporating the solvents, the nonpolar lipids were separated by developing the chromatogram with the solvent II (n-hexane:diethyl ether: acetic acid, 80:20:1.5) up to 1 cm from the top of the plate. The plates were dried under a stream of air and sprayed with primulin (Sigma, St Louis, MO, USA) solution (50 µg/mL in acetone :water, 80:20). The plates were dried again, and fluorescent lipid spots were scanned on a UV transilluminator (SL-20, Seoulin Scientific Co., Seoul, Korea) equipped with a camera (PC 1210, Canon Inc., Tokyo, Japan). The densities of lipid spots were estimated using Gel-Pro Analyzer 4.0 software (Media Cybernetics, Silver Spring, MD, USA). Standard lipids were obtained from Sigma: cholesteryl palmitate (CP; C6072), palmitate (PA; P5586), cholesterol (CL; C8667), cardiolipin (CA; C0563), 1,2-diacyl-sn-glycero-3-phospho-L-serine (PS; P7769), L-α-lysophosphatidylcholine (PC; L4129), and 3-snlyso phosphatidyl ethanolamine (PE; L4754).
Western blot analysis and antibodies
Cells were washed twice with PBS and lysed with lysis buffer (50 mM Tris-Cl, pH 7.5, 0.1 M NaCl, 1 mM ethylene diaminetetraacetic acid, 1% [v/v] Triton X-100, 10 µg/mL each of aprotinin and leupeptin, and 1 mM phenyl methan sulfonyl fluoride. A portion (30 µg) of the lysate was applied to Western blot analysis as previously described [10]. Antibodies against SREBP1 (cat. 557036) and SREBP2 (cat. 557037) were purchased from BD Biosciences (San Jose, CA, USA), and antibody for α-tubulin (Ab-1) was from Oncogene (Boston, MA, USA). Antibodies against FAS, ACC, and ACL were kindly provided by Prof. Kyung-Sup Kim (Yonsei University, Seoul, Korea). Antibodies against phosphor-GSK3α (pGSK3α, cat. 9337) and phosphor-GSK3β (pGSK3β, cat. 9336) were purchased from Cell Signaling (Beverly, MA, USA). Antibodies for GSK3 (sc-7291) and p21 (sc-397) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibody against Ab-1 (Oncogene) was used as the primary antibody. The expression level of each protein was estimated by comparing the band intensities exposed on X-ray film using Gel-Pro Analyzer 4.0 software.
Introduction of siRNA into cells
Oligonucleotides for GSK3α siRNA (5'-GAAAGACGAGCUUUACCUATT-3'), GSK3β siRNA (5'-CUCAAGAACUGUCAAGUAATT-3'), SREBP1 siRNA #1 (5'-GGCCGAAGGCAGUGCAAGAT-T-3'), SREBP1 siRNA #2 (5'-CCGAAGGCAGUGCAAGAGATT-3'), ACL siRNA #1 (5'-CAAAAUAUGUGGAGUAGUATT-3'), ACL siRNA #2 (5'-UGAAAUG-UCUUGUGUCUUATT-3'), and negative control siRNA (5'-UAGCGACUAAACACAUCAA-3') were produced by Samchully Pharm Co. (Seoul, Korea). siRNA duplexes were introduced into cells using Oligofectamine (Invitrogen, Grand Island, NY, USA) according to the manufacturer's instruction.
Senescence-associated β-galactosidase assay
Senescence-associated β-galactosidase (SA-β-gal) activity was assayed at pH 6.0 as described previously [10]. Briefly, cells were washed twice with PBS, fixed to plates by 3.5% formaldehyde for 5 minutes, and then incubated overnight in freshly prepared staining solution (40 mM citrate-phosphate buffer, pH 6.0, containing 1 mg/mL of 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside [X-gal, Sigma], 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, and 2 mM MgCl2). The stains were visible 24 hours after incubation at 37℃. By counting the numbers of the blue-stained and total cells using Image J software (NIH, Bethesda, MA, USA), percentage of SA-β-gal positive cells was obtained to estimate the degree of cellular senescence.
RESULTS
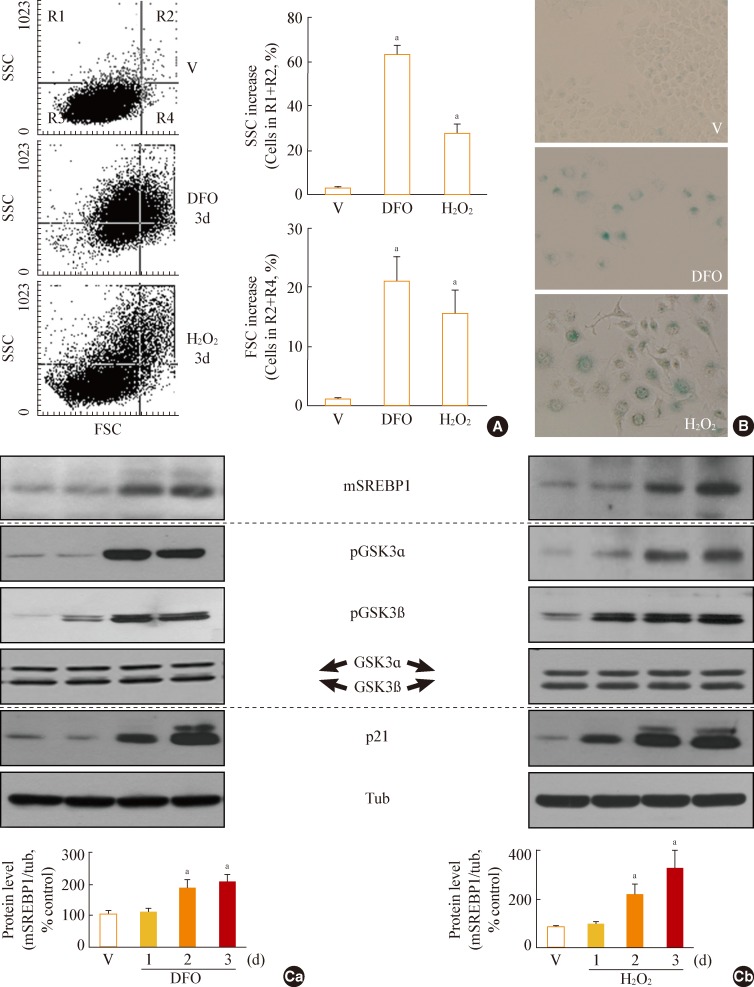
GSK3 phosphorylation (inactivation) occurs prior to increased mSREBP1 levels during stress-induced senescence
When Chang cells were exposed to 200 µM DFO or 200 µM H2O2, three typical senescence markers-increases in cell size, cellular granularity, and SA-β-gal activity-were clearly observed (Fig. 1A, B). Next, we monitored the expression profile of mSREBP1 and the phosphorylation status of GSK3 during stress-induced senescence. Phosphorylation of GSK3 increased before the increase in mSREBP1 levels in cells in which senescence was induced by either DFO or H2O2 (Fig. 1C). These results imply that GSK3 inactivation may lead to increased mSREBP1 protein levels by blocking the phosphorylation and degradation of mSREBP1. We further examined activation (phosphorylation) status of AKT and S6K to elucidate common upstream modulator of GSK3 in these senescence systems. However, no common upstream signal to inactivate GSK3 was found in the two senescence systems (data not shown), implying that diverse signals activated by different stimuli may converge on GSK3 phosphorylation (inactivation).
Pharmacological inhibition of GSK3 increases levels of mSREBP1 protein and lipogenesis
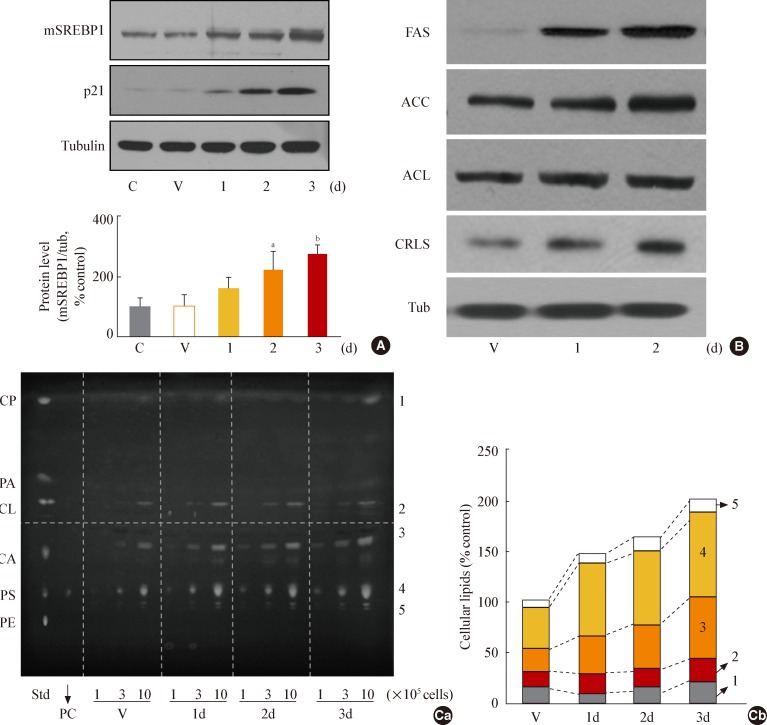
To investigate whether GSK3 inactivation leads to increased mSREBP1 expression during senescence, we employed SB41528, a selective pharmacological inhibitor of GSK3 [33]. GSK3 inhibition by SB415286 was associated with increased mSREBP1 expression (Fig. 2A) and induction of the SREBP1-targeting lipogenic enzymes FAS, cardiolipin synthase, ACC, and ACL (Fig. 2B).
To further evaluate whether GSK3 inhibition enhances cellular lipogenesis, we extracted total cellular lipids and separated them using TLC. As expected, the amount of total lipids (sum of the lipids indicated by numbers 1 to 5) (Fig. 2C) increased progressively during SB415286-induced senescence; on day 3, they were roughly twice as many compared to baseline measurements. Potential membrane lipids (sum of the lipids indicated by numbers 2 to 5): PE, phosphatidyl serine [PS], CA, and CL) (Fig. 2C) comprised approximately 82% of the total lipids in control cells and increased by 179% in SB412586-induced senescent cells; over the same period, the amount of potential storage lipids (indicated by number 1, CP) (Fig. 2C) increased minimally. These results indicate that GS K3 inhibition enhances cellular lipogenesis through increased mSREBP1 levels and transcriptional activity.
GSK3 inhibition leads to increased organellar mass and eventually induces senescence in Chang cells
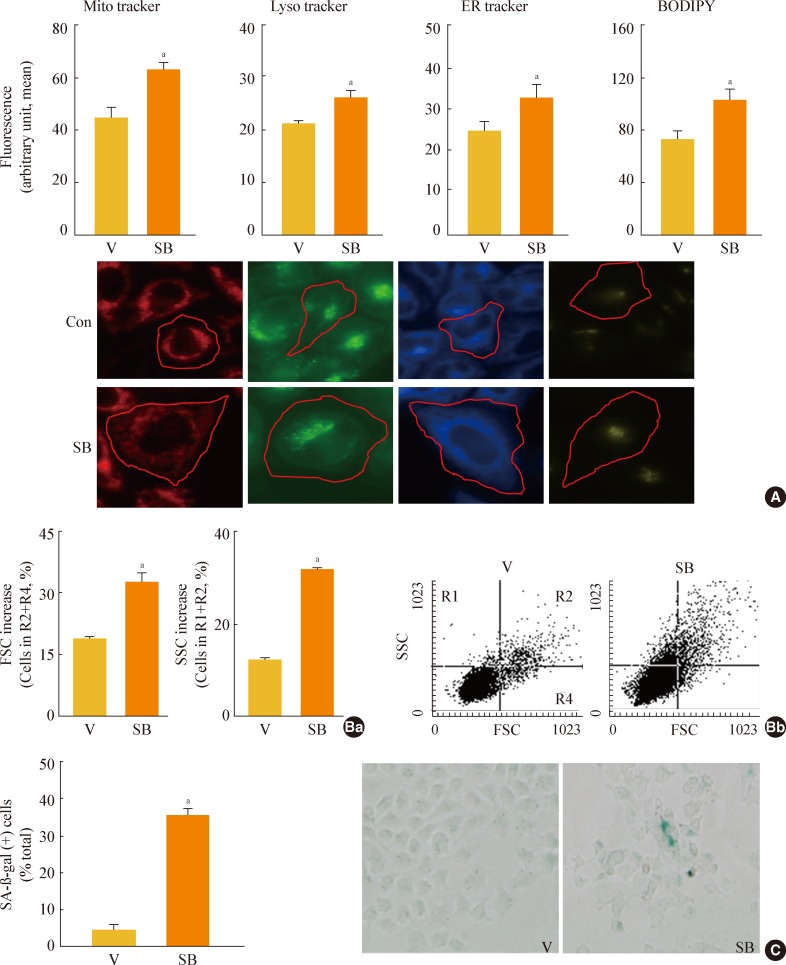
As shown by the results in Fig. 2, GSK3 inhibition led to an increase in membrane lipids that was greater than the increase in storage lipids. Therefore, we next examined whether the increase in membrane lipid production contributed to the formation of cell organelles. When we monitored the mass of membranous organelles by flow cytometry after staining cells with organelle-specific fluorescence dyes, we found that levels of most intracellular organelles, including mitochondria, lysosomes, ER, and Golgi, increased significantly during GSK3 inhibition-mediated senescence (Fig. 3A). We also observed enlarged cellular morphology, one of the characteristic signs of senescence, in SB415286-induced senescent cells in all of the images of cells incubated with organelle-specific stains (Fig. 3A, lower panels). We eventually confirmed that GSK3 inhibition by SB415286 induced senescence of Chang cells, as shown by increases in cell size and cell granularity and the presence of SA-β-gal activity (Fig. 3B, C).
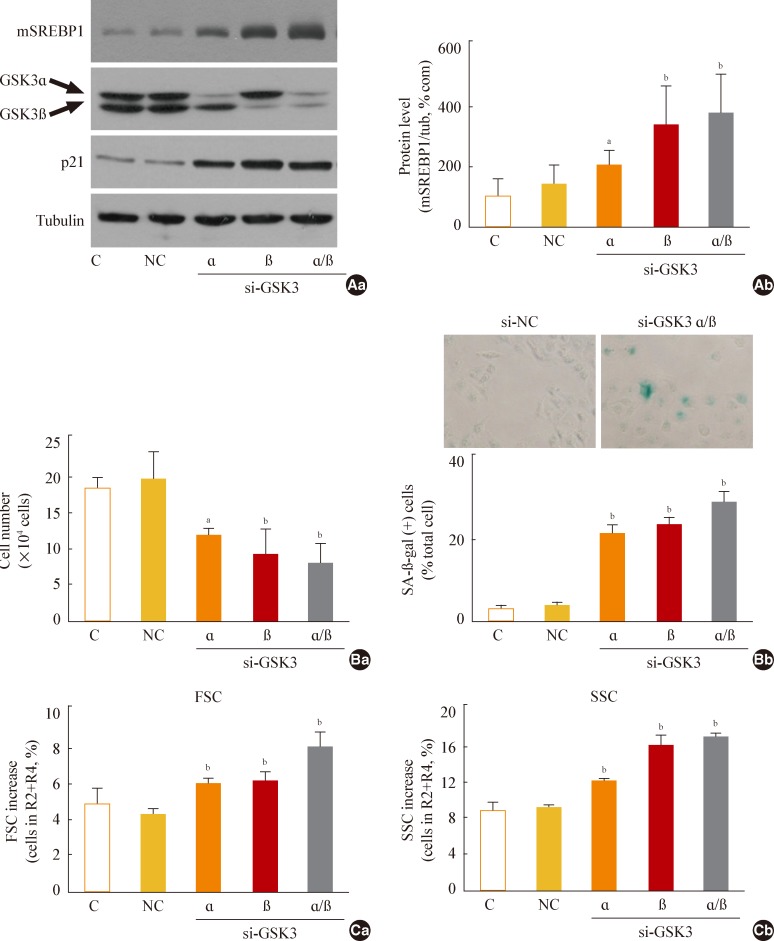
Next, we further examined whether specific knockdown of GSK3α and β would increase mSREBP1 expression and induce senescence. When we suppressed GSK3α and β expression by siRNA-mediated knockdown, we observed a significant increase in mSREBP1 levels; a delay in the cell growth rate; and senescence phenotypes, such as increases in cell size and cell granularity and gain of SA-β-gal activity (Fig. 4). These data confirm that GSK3 inactivation is linked to cell senescence, along with the accompanying enhanced lipogenesis and increased organellar mass.
SREBP1-mediated lipogenesis is critically involved in GSK3 inhibition-induced senescence of Chang cells
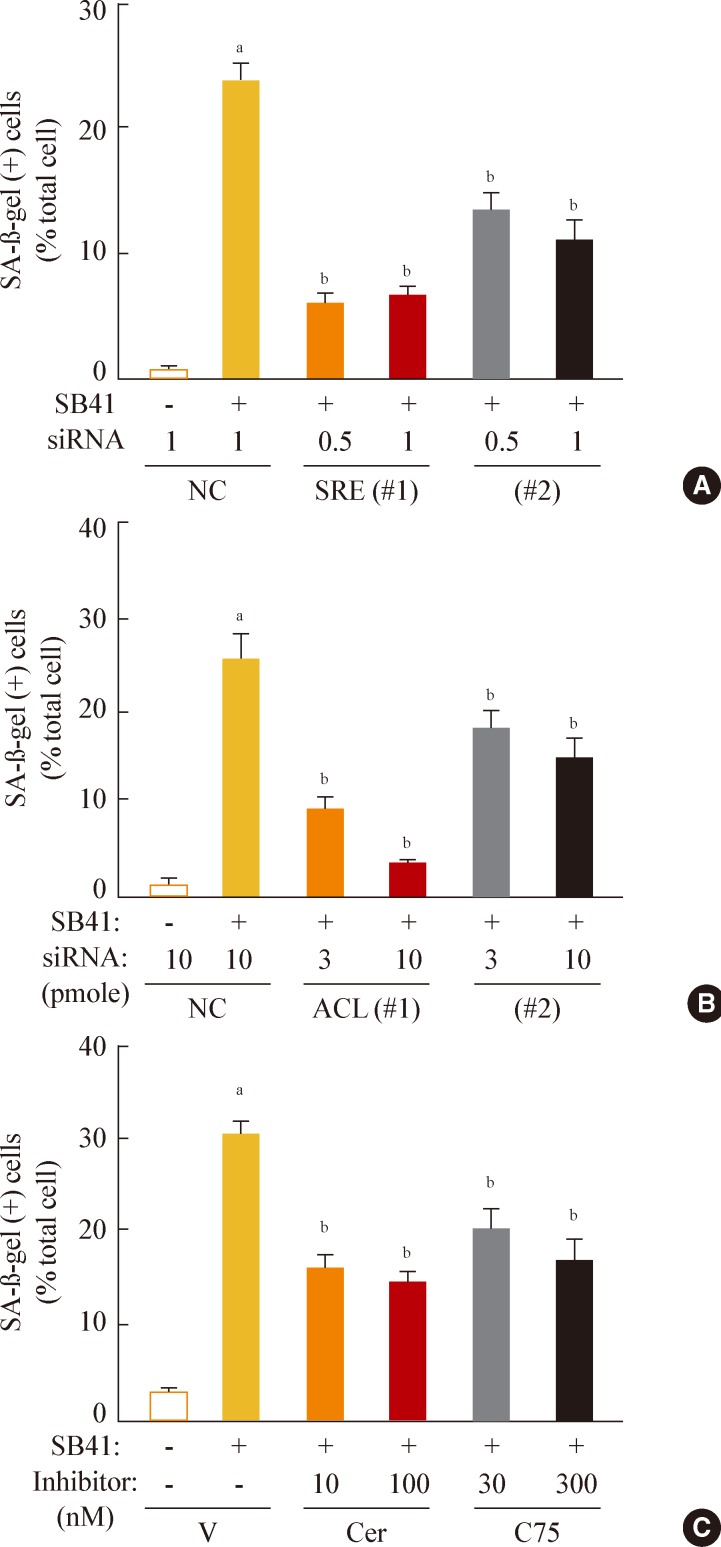
Finally, we investigated whether inhibition of lipogenesis could reverse GSK3 inhibition-induced senescence. Knockdown of SREBP1 or ACL expression effectively attenuated the SA-β-gal activity induced by GSK3 inhibition (Fig. 5A, B). Moreover, inhibition of lipid synthesis with the FAS inhibitors cerulenin and C75 [34,35] also attenuated GSK3 inhibition-induced senescence (Fig. 5C). These data demonstrate that GSK3 inactivation is an upstream event that induces cellular senescence in Chang cells via SREBP1-mediated lipogenesis. In addition, our results also show that the axis of GSK3/SREBP1/lipogenesis is associated with increased organellar mass, an important indicator of senescence.
DISCUSSION
In this study, we clearly demonstrate that inactivation of GSK3 is critically involved in enhanced lipogenesis mediated by SREBP1 and thereby contributes to the increase in organellar mass that is a typical characteristic of senescence. We also found that GSK3 inactivation induces the senescence of Chang cells through SREBP1-mediated lipogenesis. Our results confirm the role of GSK3 as a key metabolic controller that is closely associated with the metabolic phenotype of senescence, and they suggest a novel molecular background for aging and aging-related pathophysiology.
We previously reported that GSK3-mediated glycogenesis is critically involved in cell senescence [23]; in the present study, we confirmed the role of GSK3 inactivation in lipogenesis-associated cell senescence. Both glycogenesis and lipogenesis are cellular anabolic processes, but their cellular functions are quite different: the former process stores energy, and the latter generates new cellular components. Glycogenesis may occur as the result of minimal use of energy due to the irreversible arrest of cell growth in senescence, while lipogenesis may be a passive cellular defense that is used to maintain the total number of normal cellular organelles in response to organellar damage by senescent stress. However, it is also clear that enhancement of both glycogenesis and lipogenesis act as major contributors of senescence, respectively, as evidenced by the facts that knockdown of glycogen synthase blocked glycogenesis-associated senescence induced by GSK3 knockdown [23] and overexpression of mSREBP1 alone induced senescence [10]. Therefore, these metabolic phenotypes may be double-edged swords for senescence, acting as both a cause of senescence (metabolic stressor) and a consequence of senescence (defensive response).
There is another important question that remains to be answered: why does senescence enhance such different anabolic processes through inactivation of the single kinase GSK3? Moreover, a recent study suggested yet another role for GSK3 in cell functioning, in which GSK3 inactivation provokes and maintains oxidative stress by triggering mitochondrial respiratory defects [36]. Together, these results imply that GSK3 is a common regulator that activates anabolic processes and inhibits catabolic processes in mitochondria, and they emphasize the role of GSK3 as a metabolic switch to control cell senescence.
GSK3 has long been known as a negative regulator of glycogen synthesis in response to insulin [17,18]. In addition, GSK3 inhibition was reported to improve insulin resistance by enhancing insulin action in rat skeletal muscle [37], and GSK3 inhibitors, including SB415286, have been developed as drugs for the disease [33]. Contrary to our results, these reports support the protective role of GSK3 inactivation in insulin resistance, an aging-associated disease. On the other hand, an insulin or insulin-like signaling cascade, involving IGF-1 receptor, IRS, PI3K, and AKT, has also been implicated in the control of organismal lifespan and senescence [25,38,39], well supporting that GSK3, one of the major downstream targets of the insulin signal cascade, may participate in senescence. Therefore, at the present stage, it seems that GSK3 has two opposing actions in modulating senescence and alleviating insulin resistance. Nevertheless, one may argue that the concentration (7.5 µg/mL) of SB415286 used in our study was so high that cells were placed in a stress condition. In our previous study, we repeatedly applied a low dose of SB415286 (3.6 µg/mL) to mimic the effect of continuous daily application of the drugs in vivo [23]. Three repeated daily applications with the low dose SB415286 were enough to induce senescence phenotypes, whereas a single treatment of low dose (3.6 µg/mL) SB 415286 did not. Collectively, our results may raise awareness of employing GSK3 modulation in therapeutic approach of insulin resistance.
In conclusion, our results provide support for an important role for GSK3 in the modulation of SREBP1-mediated lipogenesis and senescence and in the regulation of increases in cellular mass, a phenotype typical of senescence. However, details of the mechanism by which GSK3 controls SREBP1 activity remain unclear, although a recent study indicated that phosphorylation-mediated ubiquitination is involved in the control of SREBP1 activity by GSK3, and that FBw7 is a ubiquitin ligase for SREBP1 [29]. We are currently exploring whether the senescence-associated increase in mSREBP1 expression is the result of blocking Fbw7-mediated ubiquitination or whether it is the result of increased transcriptional activation and/or activated maturation.
 XML Download
XML Download