

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Acromegaly is not a common disease and its annual estimated incidence is 3-4 cases per 1 million people worldwide [1]. Hypopituitarism is reported to occur in over 50% of patients with acromegaly 5-10 years after radiation therapy [2].
Aplastic anemia is a clinical condition that results from a marked diminution of marrow blood cell production. Its diagnosis is usually based on the presence of pancytopenia accompanied by hypocellular marrow without abnormal or malignant cells or fibrosis. It is sometimes related to connective tissue disease, idiosyncratic responses to certain pharmaceuticals, exposure to toxic chemicals, and systemic diseases, such as infection [3]. While hormonal deficiency is rarely thought of a cause of pancytopenia with hypocellular bone marrow, the mechanisms of hormonal effects on hematopoiesis have not yet been fully understood and there have been only a few cases with bone marrow hypoplasia and pancytopenia which were related to hormone insufficiencies. We experienced a case of an acromegalic patient with pancytopenia who developed in hypopituitarism after surgical treatment and radiotherapy but recovered completely with hydrocortisone replacement therapy.
CASE REPORT
A 28-year-old man was referred from a private clinic with a history of mandibular hypertrophy and peripheral thickening in the hands and feet, 6 years ago. His insulin like growth factor 1 (IGF-1) level increased to 1,197.6 ng/mL (normal, 114-492). Oral glucose tolerance test results revealed that growth hormone (GH) levels were not suppressed (63 ng/mL), so the patient was diagnosed with acromegaly. Pituitary macroadenoma had been found on magnetic resonance imaging (MRI) (Fig. 1A), and he underwent surgery through the transsphenoidal approach for pituitary macroadenoma removal the same year. One month after surgery, the IGF-1 level was 1,318.1 ng/mL, thyroid function tests were normal, and the prolactin level was 15.5 ng/mL (normal, 0-20). The adrenocorticotropic hormone (ACTH) level was 33.8 pg/mL (normal, 10-60) in the morning. MRI was performed 2 months after surgery and showed a remnant adenoma (Fig. 1B). Then, he underwent gamma-knife radiosurgery (GKS). After GKS, he was transferred to the endocrinology department and still had a high level of IGF-1 (1,298.7 ng/mL). Thus, we prescribed bromocriptine 2.5 mg per day for 1 month, which was changed to Sandostatin LAR (Octreotide acetate, Novatis Pharm., Basel, Switzerland) 20 mg monthly. After 8 months, the IGF-1 levels were still high, so we increased the dose of Sandostatin LAR to 30 mg monthly after using 20 mg. The IGF-1 levels finally showed a serial decrease and Sandostatin LAR dosage was tapered from 12 months ago (Fig. 2). Two months after Sandostatin LAR discontinuation, his complete blood count (CBC) test showed a slight decrease in leukocytes (3.2 × 109/L), anemia (hemoglobin, 10.8 g/dL), and normal platelet counts (167 × 109/L).
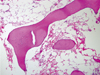
The patient visited the emergency department 6 years after radiosurgery. At that time, he presented with general weakness, diarrhea, nausea, and vomiting. On admission, his vital signs were as follows: blood pressure, 100/60 mmHg; pulse rate, 80 beat/min; and body temperature, 36.5℃. He had not been medicated during the past 4 weeks. CBC test results showed pancytopenia: leukocytes, 1.5 × 109/L; neutrophils, 0.72 × 109/L; hemoglobin, 11.0 g/dL; and platelets, 67 × 109/L. Peripheral blood smear (PBS) also revealed pancytopenia with both a low reticulocyte produrction index (0.13) and a relatively low reticulated platelet count (4.1%; normal, 0.5-5.5%). Hyponatremia was also noted: sodium, 109.9 mmol/L; potassium, 3.80 mmol/L; and chloride, 81.4 mmol/L. Hyponatremia was not corrected 2 days after hypertonic saline administration. The basal hormone status on admission, revealed cortisol deficiency-cortisol, 3.18 µg/dL (normal, 5-25) and ACTH, 17.15 pg/mL (normal, 10-60). IGF-1 was 45.78 ng/mL (normal, 114-492). Thyroid function test results were suggestive of euthyroid sick syndrome-triiodothyronine (T3), 72.3 ng/dL (normal, 80-170); free thyroxine (T4), 1.81 ng/dL (normal, 0.80-2.10); and thyroid stimulationg hormone (TSH), 3.03 µIU/L (normal, 0.3-5.0). Hypogonadism was also noted: luteinizing hormon, 0 mIU/mL (normal, 0.4-5.7); follicular stimulating hormone, 1.8 mIU/mL (1.5-12.4); and testosterone, 0.45 ng/mL (normal, 2.5-18.3). Combined anterior pituitary function tests demonstrated hypopituitarism in multiple axes except for TSH. MRI of the pituitary gland revealed an empty sella (Fig. 1D). He was administered oral hydrocortisone (10 mg) daily from the fourth admission day onward. Hypertonic saline was infused until the fifth admission day and was discontinued when the sodium level was corrected to 120.2 mmol/L. The sodium level was further corrected to 130.4 mmol/L on the third day of hydrocortisone replacement therapy. The number of leukocytes also increased to 4.36 × 109/L on the fifth day of the therapy. Because neutropenia continued for 3 years at the level of 1.1-1.5 × 109/L, he underwent bone marrow biopsy which revealed a hypocellular marrow (Fig. 3).
Five months after discharge, the patient visited the emergency department again due to general weakness, diarrhea, nausea, and vomiting. He reported that he had not taken any medications, including hydrocortisone after discharge from the hospital. Complete blood cell counts showed pancytopenia: leukocytes, 1.18 × 109/L; neutrophils, 0.86 × 109/L; hemoglobin, 9.1 g/dL; and platelets, 106 × 109/L. On PBS, normocytic anemia and neutropenia was noted, the reticulocyte production index was 0.21, mean cell volume was 82 fL (normal, 80-94), and mean cell hemoglobin was 28.5 pg (normal, 27-32). The iron profiles were as follows: iron, 50 µg/dL (normal, 80-200); total iron binding capacity, 216 µg/dL (normal, 252-456); and ferritin 327.2 ng/mL (normal, 15-332). Vitamin B12 and folate levels were normal: 856.12 pg/mL (normal, 200-950) and 7.04 ng/mL (normal, 3-17), respectively. The serum erythropoietin level was 38.2 mIU/mL (reference, 3.5-16.2). Hyponatremia and hypokalemia were also noted: sodium, 118 mmol/L; potassium, 3.45 mmol/L. To evaluate hyponatremia, we measured serum osmolarity which was 251 mOsm/kg and urine osmolarity which was 231 mOsm/kg. These results were suggestive of hypotonic hyponatremia with dysfunction of urinary concentration. Biochemical tests revealed normal liver and kidney functions. In addition, C-reactive protein (CRP) and procalcitonin were measured to rule out infection: CRP, 0.51 mg/dL (normal, 0-0.5); procalcitonin, 0.06 ng/mL (normal, ≤ 0.05). Stool test results obtained at 2 weeks were negative for leukocytes and cultures. In addition, abdominal computed tomography was normal.
In order to ascertain a role of pituitary hormone on pancytopenia, basal pituitary hormone tests were performed. Serum cortisol levels were lower than reference values-4.42 µg/dL at 8:00 AM and 4.38 µg/dL at 4:00 PM (normal, 5-25). Thyroid function test results were normal: T3, 71 ng/dL; free T4, 1.21 ng/dL; and TSH, 2.42 µIU/L. The testosterone level was 0.4 ng/mL (normal, 2.45-18.3). Combined anterior pituitary stimulation tests demonstrated hypopituitarism in all axes except for TSH (Table 1).
Again, the patient received replacement therapy with oral hydrocortisone 10 mg daily. One week after hydrocortisone replacement, the number of hemocytes increased: leukocytes, 3.17 × 109/L; hemoglobin, 9.2 g/dL; and platelets, 316 × 109/L. The serum sodium level was 137.1 mmol/L, which was in the normal range. He was restored to a good general condition. He was prescribed hydrocortisone (10 mg) daily and discharged from the hospital. His last laboratory results were almost within the normal range 3 month after hydrocortisone replacement therapy: sodium, 141.7 mmol/L; potassium, 4.19 mmol/L; leukocytes, 3.76 × 109/L; hemoglobin, 12.2 g/dL; and platelets, 258 × 109/L (Fig. 4).
DISCUSSION
Pancytopenia rarely occurs in bone marrow failure syndrome secondary to hypopituitarism. There have been several reports of pancytopenia that are related to hormone deficiency. We reviewed nine cases by using Medline search (Table 2). Six of these cases were associated with Sheehan syndrome [4-9], one was associated with macroprolactinoma [10], and one with hypothalamic glioma and one with suprasellar germinoma [11,12]. To the best of our knowledge, this is the first case of pancytopenia secondary to hypopituitarism.
It is not clear which hormones are important to induce or improve pancytopenia in cases of pancytopenia due to hypopituitarism. Most patients recover by simultaneous replacement of cortisol and thyroid hormone [4-7,9]. Lee [12] described a case of an 11-year-old girl who developed postablative hypothyroidism and prolonged pancytopenia after radiotherapy for a suprasellar germinoma while receiving cortisol replacement. Hence, the authors believed that hypothyroidism was a major contributing factor to pancytopenia in patients with panhypopituitarism. Conversely, Laway et al. [8] reported a patient with Sheehan's syndrome who had pancytopenia and hyponatremia. The patient was treated with hydrocortisone and showed a complete recovery from pancytopenia. They concluded that glucocorticoids play a crucial role in reversing pancytopenia in Sheehan's syndrome [8]. Badawi et al. [11] reported a young male patient with long-term panhypopituitarism and pancytopenia attributable to poor adherence to androgen replacement therapy, which resolved after institution of testosterone therapy. They emphasized the importance of testosterone in the treatment of hypopituitarism-induced pancytopenia.
Even though hormonal effects of hormone levels on myelopoiesis and thrombopoiesis are not clear, erythropoiesis is affected by metabolic needs and pituitary hormones play an important role in the treatment of erythropoiesis. A previous study reported that anemia occurred in 32% of patients with hypopituitarism and that the minimum recorded hemoglobin level was 9.3 g/dL [13]. Laway et al. [14] demonstrated that, anemia, leukopenia, thrombocytopenia, bicytopenia, and pancytopenia were significantly higher in patients who were diagnosed with Sheehan's syndrome than in those who were not. Among pituitary hormones, thyroid hormone and testosterone have been shown to significantly stimulate erythroid progenitors, increase erythropoietin production and potentiate erythropoietin action [15,16]. Steroid hormones directly stimulate erythropoiesis [13]. In addition, GH and IGF-1 are known to have a direct effect on erythroid and myeloid precursor progenitor cells [17,18].
Our patient showed mild pancytopenia for 3 years and pancytopenia worsen during two events of electrolyte imbalance. During each event, the patient had the similar symptoms, such as nausea, vomiting and diarrhea, and the symptoms improved with physiologic doses of hydrocortisone. Thus, he was diagnosed as having hypopituitarism with adrenal crises, which was probably caused by the stress of gastroenteritis. Uniquely, the patient showed insufficiencies of pituitary hormones except thyrotropin. Even testosterone level was low, though it had been low since he diagnosed acromegaly. We ruled out testosterone deficiency because testosterone levels had been low for a long time and there were no symptoms associated with testosterone deficiency. At first, we recommended testosterone replacement therapy, but he refused it. Moreover, after only administration of hydrocortisone, the patient showed a full recovery from pancytopenia. Therefore, this case supports the hypothesis that glucocorticoids are effective in treating pancytopenia induced by hypopituitarism.
In conclusion, hypopituitarism itself might be a possible cause of pancytopenia, although it rarely develops the aforementioned hematologic abnormalities due to an excess of GH in acromegaly. Physicians should consider hormone insufficiency a cause of pancytopenia in patients who have undergone prior surgical treatment or radiotherapy for pituitary gland lesions.




 XML Download
XML Download