

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The association metabolic disturbances and androgen excess in women has been recognized since at least the 18th century [1,2]. In the 1940s, Vague [3] noted that women with male-pattern or android body fat distribution were at increased risk for diabetes and cardiovascular disease (CVD). Rare syndromes of insulin resistant diabetes, hyperandrogenism and acanthosis nigricans were formally described in 1976 [4]. Evaluation of the cellular and molecular defects in insulin action in these syndromes resulted in the identification of mutations in the insulin receptor gene altering its number, binding or function [5,6]. In a subset of these disorders also associated with partial or total lipoatrophy, several Mendelian disorders resulting from mutations in genes regulating adipocyte differentiation or architecture were identified [6,7]. The common feature of these molecularly diverse disorders was hyperinsulinemia implicating insulin in the pathogenesis of the reproductive disturbances [8]. The recognition that women with polycystic ovary syndrome (PCOS) had basal and glucose-stimulated hyperinsulinemia, independent of obesity [9], and acanthosis nigricans [10,11] suggested insulin might also be important in the pathogenesis of this common syndrome of unknown etiology [8].
PCOS is one of the most prevalent endocrine disorders of reproductive age women, affecting approximately 7% of this population [12-14] or to 4 million women ages 15-44 years in the US alone using 2010 Census population estimates. Worldwide PCOS prevalence rates are similar, except in Latinas where they may be even higher [8]. PCOS is diagnosed by its reproductive phenotype of hyperandrogenism, chronic anovulation, and polycystic ovaries (PCO) [8]. It is frequently associated with substantial insulin resistance, pancreatic β-cell dysfunction and obesity [8,15,16]. PCOS is a leading risk factor for metabolic syndrome (MetS) [17-20], a constellation of CVD risk factors associated with visceral adiposity and insulin resistance [21], and type 2 diabetes mellitus (T2D) in adolescent [18, 22] as well as in adult women [23-25]. Women with PCOS appear to be at risk for a number of other conditions associated with insulin resistance, including gestational diabetes, preeclampsia, sleep apnea and non-alcoholic fatty liver disease [26]. Women with epilepsy and bipolar disorder have an increased prevalence of PCOS, independent of medications [27,28].
The metabolic sequelae of PCOS persist after menopause, particularly the increased T2D risk [29-31]. Moreover, several studies of postmenopausal women with features of PCOS suggest that they are indeed at increased risk for CVD [32,33], as would be expected from the increased prevalence of CVD risk factors including obesity, dysglycemia, MetS, T2D, elevated low-density lipoprotein (LDL) levels, and endothelial dysfunction in PCOS [34]. PCOS is highly heritable [35,36] and male as well as female relatives, including infants and children, have metabolic and reproductive phenotypes [35,37-40].
Despite more than 75 years of investigative effort, the etiology(s) of PCOS remain unknown [8]. However, major advances in understanding PCOS have been made in the last 25 years through investigation of the links between its reproductive and metabolic abnormalities. Most importantly, the insight that insulin resistance and the resultant hyperinsulinemia contribute to the reproductive phenotype of PCOS has led to a major new therapeutic modality with insulin-sensitizing agents [15]. However, insulin resistance does not account entirely for the reproductive features of the syndrome. The role of androgens in the pathogenesis of PCOS has been reexamined in recent years since prenatal androgen exposure can create an almost complete phenocopy of PCOS in non-human primates [41]. The well-known familial aggregation of PCOS as well as twin studies indicate that there is also a genetic susceptibility to the disorder [35-37]
REPRODUCTIVE PHENOTYPE IN PCOS
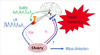
The biochemical reproductive phenotype in PCOS is characterized by increased luteinizing hormone (LH) relative to follicle-stimulating hormone (FSH) secretion and hyperandrogenism [42]. There is increased frequency of gonadotropin-releasing hormone (GnRH) secretion that selectively increases LH release, while simultaneously suppressing FSH secretion (Fig. 1) [42]. Pituitary sensitivity to GnRH is also increased, which appears to be estrogen-mediated [42]. Increased LH levels stimulate increased ovarian theca cell androgen production [43]. PCOS theca cells secrete increased androgens, both basally and in response to LH [44].
Acyclic FSH secretion results in arrested ovarian follicular development so that granulosa cells do not develop sufficient aromatase capacity to completely aromatize androgens into estrogens [43]. Often, there is increased adrenal androgen production (Fig. 1) [45]. Androgens play an essential role in the pathogenesis of the increased LH release by causing insensitivity to the normal feedback effects of estrogen and progesterone feedback to slow GnRH frequency in PCOS (Fig. 1); normal feedback can be restored by androgen receptor blockade [46]. Hyperinsulinemia secondary to insulin resistance plays a pivotal role in amplifying both the steroidogenic as well as the gonadotropin secretory abnormalities [8].
There are intrinsic abnormalities in PCO. In both ovulatory and anovulatory PCO, the proportion of early growing (primary) follicles is significantly increased with a reciprocal decrease in the proportion of primordial follicles compared to normal ovaries [47]. These differences are particularly striking in anovulatory PCO [47]. There is decreased atresia of follicles from PCO in culture compared to those from normal ovaries [47]. Theca cells from PCO have constitutive increases in activity of multiple steroidogenic enzymes [44]; it is postulated that a similar defect contributes to adrenal androgen excess in affected women [44,45].
INSULIN RESISTANCE IN PCOS
Women with PCOS have defects in both peripheral, which reflects primarily skeletal muscle, and hepatic insulin action, as well as pancreatic β-cell dysfunction [8,48]. These abnormalities are independent of obesity, although obesity substantially worsens these parameters [8,15,49]. However, in certain populations of women with PCOS, such as those from Scandinavia, obesity per se seems to account for insulin resistance [50,51]. This finding suggests that there may be ethnic variations in the mechanism of insulin resistance in PCOS.
The molecular mechanisms of insulin resistance in PCOS differ from those in other common insulin resistant states, such as obesity and T2D [8]. Despite the phenotypic similarity between PCOS and the type A syndrome of extreme insulin resistance and hyperandrogenism [5,8], no mutations in the coding portion of insulin receptor gene have been found in PCOS [8]. Studies in isolated subcutaneous adipocytes have shown no differences in insulin receptor number or affinity compared to weight-comparable control women [49,52]. However, the presence of post-binding defect in insulin signaling in adipocytes of women with PCOS was suggested by significantly decreased sensitivity to insulin-mediated glucose transport [49,52]. The abundance of GLUT4 glucose transporters was decreased in one [53] but not in another study [54] of isolated subcutaneous adipocytes from affected women. Additionally, an enhanced lipolytic effect of catecholamines was found in isolated visceral adipocytes in PCOS, which could contribute to insulin resistance by increasing free fatty acids release directly into the portal circulation [55]. The opposite abnormality, resistance to catecholamine-induced lipolysis, was present in isolated subcutaneous abdominal adipocytes [56].
Constitutive increases in insulin receptor serine phosphorylation were found in receptors isolated from cultured skin fibroblasts and from skeletal muscle, a classic insulin target tissue, in PCOS [57]. The increase in insulin receptor serine phosphorylation was accompanied by a decrease in insulin receptor tyrosine kinase activity that is essential for normal insulin signal transduction [8,57]. Studies using serine kinase inhibitors have suggested that a serine kinase extrinsic to the receptor is responsible for the constitutive increase in insulin receptor serine phosphorylation [58]. Studies in vivo have demonstrated a post-receptor defect in IRS-1-mediated activation of phosphatidylinositol 3-kinase (PI3K) activity, an early step the insulin signaling cascade, in skeletal muscle biopsies in parallel with decreased insulin-mediated glucose uptake in PCOS [59]. This study supports a physiologically relevant role for defective insulin signaling in PCOS, since skeletal muscle is the major site of insulin-mediated glucose uptake [60].
Defects in insulin signaling persist in myotubes cultures established from skeletal muscle biopsies obtained in women with PCOS, including decreased IRS-1-associated PI3K activity [61], analogous to findings in skeletal muscle biopsies during in vivo euglycemic hyperinsulinemic clamp studies in affected women [59]. Decreased IRS-1-mediated PI3K activity is associated with increased phosphorylation of IRS-1 on serine 312, a regulatory site that inhibits signaling, in PCOS myotubes [61]. These observations suggest that increased serine phosphorylation of IRS-1 may contribute to insulin resistance in the major target tissue for glucose uptake [59,61].
In two studies, however, PCOS myotubes cultures established from affected women with documented insulin resistance in vivo [61,62] were no longer insulin resistant. Indeed, Corbould et al. [61] found that PCOS myotubes have increased basal and insulin-stimulated glucose transport. Increased abundance of the non-insulin regulated GLUT1 glucose transporter accounted for the increased rates of basal glucose uptake. In contrast, decreased insulin-stimulated glucose transport persisted in another study of PCOS myotubes [54] from insulin resistant affected women. The reason for the discrepancies between these studies in PCOS myotubes is unclear. Nevertheless, these observations suggest that both intrinsic abnormalities and extrinsic factors in the in vivo environment account for insulin resistance in PCOS myotubes [61].
The insulin resistance in skin fibroblasts [63] and skeletal muscle [64] in PCOS is selective, affecting metabolic, but not mitogenic signaling pathways. Further, there appear to be tissue-specific differences in insulin sensitivity in PCOS. Insulin action to stimulate androgen production is preserved in ovarian theca cells [43], while granulosa-lutein cells are resistant to insulin's action on glucose metabolism [65]. These findings may explain the paradox of the persistent reproductive actions of insulin in the face of metabolic insulin resistance in PCOS [8].
In skeletal muscle, there is intriguing evidence that activation of mitogenic insulin signaling pathways contributes to resistance to insulin's metabolic actions in PCOS. Both in skeletal muscle biopsies acutely isolated from the in vivo environment [64,66] as well as in long-term cultures of myotubes established from these biopsies [64], there is constitutive activation of the mitogen-activated protein kinase (MAPK)-extracellular signal-regulated kinase (ERK) pathway in PCOS. Raf-1 abundance is increased and the activity of p21Ras is decreased suggesting that altered mitogenic signaling begins at the level of Raf-1. Pharmacologic inhibition of MEK1/2 reduces IRS-1 Serine 312 phosphorylation and increases IRS-1 association with the p85 subunit of PI3K in both PCOS and control women. This observation suggests that MAPK-ERK pathway contributes to the normal feedback inhibition of metabolic insulin signaling through the serine phosphorylation of IRS-1. Furthermore, constitutive activation of this serine kinase mitogenic pathway may contribute to metabolic insulin resistance in PCOS [64].
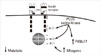
Serine phosphorylation of a key regulatory enzyme of androgen biosynthesis, cytochrome P450c17, which catalyzes both 17α-hydroxylase and C17,20 lyase activities, increased its C17,20 lyase activity [67]. This observation has led to the hypothesis that the same serine kinase contributes to insulin resistance through serine phosphorylation of the insulin receptor and, perhaps, IRS-1, as well as hyperandrogenism through serine phosphorylation of P450c17 [8,68]. However, attempts to prove this hypothesis have been unsuccessful to date [69]. Recently, the Rho-associated, coiled-coil containing protein kinase/Rho pathway was identified as a candidate pathway that can serine phosphorylate both P450c17 and the insulin receptor (Fig. 2) [70].
RELATIONSHIP BETWEEN ANDROGENS AND INSULIN
It has been long debated whether hyperandrogenemia causes insulin resistance or vice-versa. Studies in which insulin levels were lowered by diazoxide [71] or insulin sensitivity was improved by metformin [72] or the thiazolidinedione, troglitazone [73], showed that insulin can directly stimulate ovarian androgen production in PCOS. This action appears to be mediated by insulin acting through its cognate receptor [74] rather the spillover occupancy of IGF-1 receptors as previously thought [8]. However, this action is only seen at more physiologic insulin levels in the presence of LH suggesting that insulin acts more as a co-gonadotropin [43]. It has also been shown that insulin can stimulate adrenal androgen production by enhancing sensitivity to ACTH [75]. Insulin does not affect ovarian function in normal women [76] suggesting that pre-existing polycystic ovarian changes, such as theca cell hyperplasia, are essential for insulin-mediated ovarian hyperandrogenism. In addition, lowering insulin levels ameliorates, but does not abolish, hyperandrogenism suggesting the coexistence of other contributing factors.
Insulin signaling in the brain is also important for the control of reproduction as well as body weight in animal models [8]. The selective central nervous system insulin receptor female knockout mouse develops disrupted LH release, impaired folliculogenesis and obesity [77]. Furthermore, targeted disruption of insulin and leptin receptors in hypothalamic and pituitary pro-opiomelanocortin neurons results in hyperandrogenemia, peripheral insulin resistance and reduced fertility in female mice [78]. Conversely, global disruption of the pituitary insulin receptor [79] protects against infertility and increases in LH release that occur in female mice with diet-induced obesity. These studies in female mice with diet-induced obesity [79,80] indicate that insulin signaling in the pituitary and ovary are preserved, despite resistance to insulin action in metabolic tissue. This finding is another example of tissue-specific differences in insulin action that may contribute to the reproductive phenotype of PCOS. The observation that insulin sensitizing drugs can restore ovulation in women with PCOS [8,81] is consistent with a role for central nervous system insulin sensitivity in the control of human reproduction, although alterations in gonadal steroid feedback or peripheral insulin levels may also contribute to the ovulation-inducing effects of insulin sensitizing drugs [81].
ANDROGENS IN THE PATHOGENESIS OF PCOS
Women with upper-body obesity, who have many features of MetS, often have increased androgen production [82,83]. Higher levels of endogenous androgens are associated with increased risk for MetS in women [84]. Androgens are independent predictors of MetS in PCOS [17,18]. It has been hypothesized that androgens are a common final path for these metabolic defects in PCOS and upper-body obesity [85].
Administration of testosterone to female-to-male transsexuals to achieve levels in the normal male range results in decreased insulin-mediated glucose uptake [86] and increased visceral fat mass [87]. In normal postmenopausal women, administration of a weak synthetic androgen also increases visceral fat [88]. Consistent with these observations, blocking androgen action with a receptor antagonist in women with PCOS can improve insulin sensitivity, visceral adiposity, and dyslipidemia [89,90]. Additional factors contribute to insulin resistance, since suppression of androgens improves but does not completely restore normal insulin sensitivity in PCOS [91, 92]. Nevertheless, these findings indicate that there are adverse and reversible metabolic actions of androgens in women.
The role of androgens in the pathogenesis of PCOS has received renewed attention over the past 15 years [35,41]. The landmark studies of Dumesic et al. [41] have shown that prenatal androgen exposure can reproduce the reproductive and metabolic features of PCOS in non-human primates. Similar effects of prenatal androgens are seen in other species, such as sheep [41]. In humans, the putative source of androgens would be the fetal ovary or adrenal since the placenta is an effective barrier to maternal androgens excess [8]. Human experiments of nature support the hypothesis that fetal androgen excess, secondary to congenital adrenal hyperplasia or androgen-secreting neoplasms, can permanently alter LH secretion [45]. Androgen levels have not been elevated in cord blood from female offspring of women with PCOS in two studies [39,93], while a third study did find increased testosterone levels using a less specific hormone assay [94]. However, these findings do not preclude a role for intrauterine androgen excess earlier in gestation as the fetal ovary does express the enzymes required for androgen biosynthesis, P450c17, as early as the second trimester [95]. It is also possible that androgen exposure at later developmental windows during childhood and puberty programs features of PCOS. Elevated testosterone levels have been found in pubertal daughters of women with PCOS [40].
EVIDENCE FOR A GENETIC SUSCEPTIBILITY TO PCOS
Evidence for a genetic susceptibility to PCOS is provided by well-documented familial clustering of PCOS, with to 40% of reproductive age sisters affected with hyperandrogenemia [35,37]. Twin studies have shown a correlation of 0.71 between monozygotic twins and 0.38 between dizygotic twins for PCOS [36] suggesting a major influence of genetic factors. Although some studies have suggested that there is an autosomal dominant mode of inheritance, these studies have been limited by a lack of prospective design, a failure to examine many first-degree relatives, and an unknown phenotype, except in reproductive age women [37]. PCOS is more likely a complex genetic disease with at least several major susceptibility genes [96].
Hyperandrogenemia is the major underlying reproductive phenotype in PCOS families and this finding has been replicated by other investigators [97,98]. There are two affected phenotypes in sisters of reproductive age: 1) classic PCOS with hyperandrogenemia and oligomenorrhea and 2) hyperandrogenemia with regular menses. Brothers of women with PCOS have elevations in the adrenal androgen, dehydroepiandrosterone sulfate [99], a marker of male androgen excess since testicular androgen production is tightly regulated by testosterone feedback on the hypothalamus [100]. This observation suggests that they have the same defect in androgen biosynthesis as their proband sisters with PCOS [99].
Affected sisters with either of the hyperandrogenemia phenotypes have insulin resistance [101], other MetS risk factors and elevated LDL levels [19]. Our studies indicate that mothers and brothers also have elevated LDL levels and evidence for insulin resistance [102]. Therefore, reproductive and metabolic abnormalities track together in PCOS families suggesting that they may reflect variation in the same gene or in closely linked genes, be causally related, or have a common pathogenesis. Since PCOS is a heterogeneous disorder, there may be genetic mechanisms other than hyperandrogenemia and non-genetic (e.g., environmental) factors that result in the PCOS phenotype. However, some of the phenotypic heterogeneity of PCOS appears to reflect variable expression of the same gene since several reproductive phenotypes can occur within family members who would be expected to share the same genetic basis for the disorder [35].
GENETIC ANALYSES IN PCOS
Most genetic analyses of PCOS have used candidate gene approaches. These are hypothesis-based studies where genes are selected because of prior evidence implicating them in disease risk [103] and, as a consequence, are limited by known biology. Given the diverse reproductive and metabolic disruptions that are features of PCOS, there is a wide range of biologic pathways from which to select candidate genes [104]. Mutational analyses of the insulin receptor gene were negative in PCOS, despite phenotypic similarity to the syndromes of hyperandrogenism and extreme insulin resistance [8]. Linkage studies implicating the gene encoding cholesterol side change cleavage enzyme, CYP11a, and the insulin gene VNTR could not be replicated in larger studies [8,37]. Case-control association studies of more than 150 candidate genes for PCOS have been limited by small sample size, phenotypic heterogeneity due to differences in diagnostic criteria, potential population stratification, confounding associated disorders such as obesity, limited examination of gene variants within each candidate gene, lack of replication and failure to control adequately for multiple testing [8,37]. Thus, the vast majority of genes implicated as associated with PCOS in these studies require further validation.
We have used the transmission disequilibrium test (TDT), a type of family-based association testing to map PCOS susceptibility variants employing a candidate gene approach [104]. The TDT tests for association in the presence of linkage using parent-affected-child trios to examine transmitted and non-transmitted parental alleles obviating the need for multiplex families [105]. We found strong evidence by TDT that an allele of a dinucleotide repeat D19S884 on chromosome 19p13.2 was linked and associated with the PCOS reproductive phenotype [37]. These findings were replicated in an independent sample of PCOS families and in a case-control study [8,37]. D19S884 is a microsatellite marker that had been selected for mapping the insulin receptor but it mapped to intron 55 of the fibrillin-3 (FBN3) gene located approximately1 Mb centromeric to the insulin receptor gene on chromosome 19p13.2 [96]. The FBN3 PCOS susceptibility allele is also associated with evidence for insulin resistance in women with PCOS and for pancreatic β-cell dysfunction in brothers, suggesting a sex difference in the associated metabolic phenotypes [96]. Fibrillin-3 shares sequence and structural similarity with fibrillin-1, which is mutated in Marfan syndrome [37,96]. Fibrillins are extracellular matrix molecules that modulate the bioavailability of members of the TGFβ signaling family. Members of this family are important in regulating reproductive and metabolic pathways [8]. The expression of fibrillin-3 is decreased in PCO [106].
The human HapMap contains common haplotypes and single nucleotide polymorphisms (SNPs) that identify these haplotypes, so-called tag SNPs [107]. Genome-wide association studies (GWAS) map the genome by genotyping these tag SNPs [107]. Population stratification can be controlled for in GWAS by the by adjusting for axes of ancestry-specific variation [8]. GWAS permit an unbiased interrogation of the entire genome for novel disease susceptibility loci and are, unlike candidate gene approaches, hypothesis generating [103]. The first PCOS GWAS [108] was in Han Chinese PCOS cases diagnosed by the Rotterdam criteria. There was strong evidence for association between PCOS and loci on chromosomes 2p16.3, 2p21 and 9q33.3. A second study by the same investigators [109] reported findings on a total sample of 8,226 Han Chinese PCOS cases and 7,578 controls confirmed the original three loci and identified eight new loci at 9q22.32, 11q22.1, 12q13.2, 12q14.3, 16q12.1, 19p13.3, 20q13.2, and a second independent signal at 2p16.3.
These loci contain several high priority candidate genes for PCOS, such as the genes encoding the LH, FSH, and insulin receptors, as well as genes associated in previous GWAS with T2D, THADA and HMGA2 [8]. As expected, new potential candidate genes for PCOS were also identified in pathways regulating transcription, chromatin structure, and cell growth, all plausible pathways for PCOS. Known genes located nearby the most significant SNP at 2p16.3 are GTF2A1L and LHCGR. GTF2A1L (TFIIA-alpha and beta-like factor) is germ cell-specific and a highly expressed in adult testis. LHCGR encodes the receptor for LH and human chorionic gonadotropin. The second SNP at 2p16.3 maps to the FSHR gene, which encodes the FSH receptor, another highly plausible candidate gene for PCOS given its characteristic abnormalities of folliculogenesis. The most significant SNP at 2p21 is in the THADA gene originally identified in thyroid adenomas but associated with T2D in a European GWAS [108]. The SNP at 9q33.3 is located within the DENND1A gene, which encodes a domain differentially expressed in normal and neoplastic cells (DENN) that can bind to and negatively regulate endoplasmic reticulum aminopeptidase-1.
The most significant SNP at 9q22.32 is located in an intron of the C9orf3 gene, which encodes C9orf3 protein, a member of M1 zinc aminopeptidase family. The SNP at 11q22.1 maps to an intron of the YAP1 gene, which encodes the YAP1 protein that is a transcriptional regulator critical in the Hippo signaling pathway. The most significant signal at 12q13.2 maps to an intragenic region between the RAB5B and SUOX genes, a region implicated in type 1 diabetes susceptibility. The most significant SNP at 12q14.3 maps to an intron of the HMGA2 gene, which encodes a transcription factor associated with a number of traits including T2D. The nearest gene to the signal at 16q12.1 is the TOX3 gene, which encodes the TOX3 protein, a member of the high-mobility-group proteins involved in chromatin structure. At 19p13.3, the most significant SNP maps to an intron of the INSR gene, which encodes the insulin receptor, a long-standing high priority candidate gene for PCOS. The most significant signal at 20q13.2 maps to an intragenic region between the SUMO1P1 and ZNF217 genes.
Two of the GWAS signals in Han Chinese PCOS were confirmed in large, well-designed studies in PCOS women of European origin that contained both discovery and replication cohorts [110,111]. The finding that the same genetic variants contribute to PCOS susceptibility in Chinese and European populations suggests that these susceptibility variants were present in an ancestral population prior to migration out of Africa [8]. Both studies replicated the association between DENND1A and PCOS [110,111]. The first study [110] also replicated the association between THADA and PCOS. The second study [111] found a significant association between one DENND1A variant and testosterone levels suggesting that this trait most likely had a genetic basis, consistent with the findings of Legro et al. [35]. Furthermore, the classic PCOS phenotype of hyperandrogenism and irregular menses but not PCO was associated another DENND1A variant [111] suggesting that there is a genetic susceptibility to the NICHD but not to the Rotterdam PCOS phenotype.
SUMMARY AND CONCLUSIONS
In summary, pathways linking metabolic cues with reproductive status are highly evolutionarily conserved being present in insects, flatworms, birds and mammals [8]. The recognition that the common reproductive disorder, PCOS, was associated with insulin resistance focused attention on elucidating the mechanisms of this association in humans [8]. This research has led to the discovery that insulin is also a reproductive hormone and that insulin signaling in the central nervous system is important for normal fertility. Moreover, androgens have adverse effects on insulin sensitivity and body fat distribution that contribute to the metabolic features of PCOS. Androgens may also play a role in the pathogenesis of PCOS through programming actions at critical developmental windows.
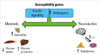
The reproductive and metabolic features of PCOS cluster in male as well as female relatives of affected women suggesting genetic susceptibility to these traits. Recent analyses using candidate gene as well as agnostic approaches have identified a number of confirmed disease susceptibility loci. These loci contain genes that are obvious high priority candidates, such as the receptors for LH, FSH, and insulin. Loci containing genes implicating new pathways in disease pathogenesis have also been mapped. In conclusion, the study of mechanisms linking reproduction and metabolism in PCOS has provided invaluable insight into novel actions of insulin and androgens, which has been translated into a new therapeutic approach with insulin sensitizing drugs. Furthermore, genetic analyses have implicated additional biologic pathways that may mediate this association (Fig. 3).
 XML Download
XML Download