

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Mixed adrenal tumors composed of more than one cell type are uncommon; they are called composite pheochromocytomas [1] and corticomedullary mixed tumors [2]. This type of pheochromocytoma is called composite or mixed, depending on whether pheochromocytoma and nonpheochromocytomatous combined components are of the same or different embryonic origin. About 50 cases of composite pheochromocytoma have been published in the English medical literature [3]. Composite pheochromocytoma is a well-defined neoplasm of the medulla and the tumor consists of both endocrine and neural components. Histologically, the endocrine portion is that of a pheochromocytoma, whereas the neural portion has been reported as ganglioneuroma, ganglioneuroblastoma, neuroblastoma, neuroendocrine carcinoma or malignant peripheral nerve sheath tumor [4]. Pheochromocytoma is usually characterized by a catecholaminergic effect with hypertension, whereas ganglioneuroma is a rare mature neuroblastic tumor that is typically nonmetabolically active, and so it is usually asymptomatic [5,6].
To date, less than 50 cases of composite pheochromocytoma have been reported [1-6], and two Korean cases of composite pheochromocytomas have been reported in the English and Korean literature [5,6]. Here, we report additional six cases of unusual tumor of the adrenal gland and the tumor exhibited a mixed phenotype composed of cells of adrenal medullary and neural lineages.
CASE REPORT
1. Clinical summary
Case 1 was a 54-year-old normotensive woman visited an endocrinology clinic of Gachon University Gil Hospital in February 2010. She previously had visited the hospital in May 2000 to evaluate incidentally found right adrenal mass. Abdominal computed tomography (CT) showed a 4 cm-sized well defined heterogeneous attenuated soft tissue mass in right adrenal gland (Fig. 1A). She did not complain any subjective symptoms and wanted to be regular followup. During 10 years of preoperative follow-up period, she experienced two episodes of headache. Case 2 was a previously healthy 33-year-old man came to Gachon University Gil Hospital in order to evaluate incidentally found adrenal mass at health screening examination. Pre contrast CT revealed an 11 cm-sized right adrenal mass with heterogenous low density, which was compressing liver (Fig. 1B). After contrast-enhancement, the mass was heterogeneously well enhanced at the peripheral solid portion. Case 3 was a 37-year-old man visited an emergency room of Samsung Seoul Hospital in July 2005 for severe headache. He had been suffered from severe headache for one year when hypertension and medication started. At that time, he also undertook a brain magnetic resonance for his severe headache. Abdominal CT taken at the emergency room showed a well-defined 5.0 cm-sized heterogeneous mass at the right adrenal gland (Fig. 1C). Case 4 was a 58-year-old man who visited an emergency room of Samsung Seoul Hospital in January 2008. He had been suffering from headache and dizziness for the previous 2 months although he had been undertaken antihypertensive drugs for the previous 4 years. Abdominal CT revealed a 3.3 cm-sized right enhancing mass with central necrosis and cystic changes at right adrenal gland (Fig. 1D). Case 5 was a previously healthy 50-year-old man. A health screening taken in August 2005 at Samsung Seoul Hospital revealed increased level of 24 hour urine vanillylmandelic acid (VMA; 7.7 mg/day) and a 3.0 cm-sized left adrenal mass on abdominal CT. Case 6 was a 64-year-old man visited an emergency room of Gachon University Gil Hospital because of a sudden onset of palpitation, limb tremor and headache for 2 days in February 2011. His blood pressure was 220/100 mmHg at that time. He had experienced two other such episodes after drinking alcohol during the previous 10 years. Abdominopelvic CT showed a 4.0 cm-sized heterogeneous enhancing mass in the right adrenal gland.
The biochemical profiles showed that the level of preoperative urine metanephrine was increased in all the 6 cases and the urine VMA level was elevated in cases 2, 3, and 6. But, endocrine profile of blood and/or urine showed increased catecholamines including epinephrine, norepinephrine or dopamine. To control the preoperative hypertension, calcium channel blockers, including bevantolol and nifedipine and alpha-antagonists were administered. All the cases underwent laparoscopic adrenalectomies. During the follow-up period ranging from 3 months to 6 years, their hypertension was controlled without any medication after adrenalectomy. The levels of blood/urine catecholamines returned to normal with no radiologic evidence of metastasis after the operation. The pre- and postoperative the 24 hour analysis of urine VMA and metanephrine were shown in Table 1. All the patients and their families were not associated with neurofibromatosis or multiple endocrine neoplasm (MEN) syndrome.
2. Pathological findings
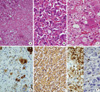
Grossly, the laparoscopically resected specimens consisted of irregular pieces of firm pinkish tan tissue in cases 2, 3, and 6. A well defined encapsulated oval shaped brownish yellow mass was found in cases 1, 4, and 5. The size of each tumor ranged from 3.0 to 11.0 cm. The cut surface was yellowish brown to mahogany-brown color (Fig. 1E, F). The specimen was received in 10% neutral-buffered formalin and processed routinely. In addition to routine hematoxylin and eosin stains, immunohistochemistry for phosphorylated neurofilament (2F11, 1:100 dilution; Dako, Glostrup, Denmark), S-100 protein (polyclonal, prediluted; Dako), synaptophysin (SY38, prediluted; Dako) and chromogranin A (DAK-A3, prediluted; Dako) were performed on formalin-fixed, paraffin-embedded tissue. Microscopically, the masses were characterized by nests, anastomosing cords and trabeculae of large polygonal cells, i.e., Zellballen pattern (Fig. 2A). Between the nests, large globoid ganglion-like cells in a fibrillary schwannian loose stroma were identified. In the portion of trabeculae of large chromaffin cells, some of them had tiny nucleoli and abundant, basophilic granular cytoplasm, with prominent intracytoplasmic inclusions (Fig. 2B). Focal melanin-like pigmentation of the chromaffin cells was also noted. Mitotic figures were rare. That portion was diagnosed as pheochromocytoma. The bundles of spindle cells were admixed with the ganglioid cells, and the findings were consistent with ganglioneuroma (Fig. 2C). The ganglioid cells were strongly positive for phosphorylated neurofilament (Fig. 2D) and S-100 protein, while the large chromaffin cells were positive for synaptophysin (Fig. 2E) and chromogranin A with admixed S-100 protein-positive sustentacular cells (Fig. 2F). The tumors were diagnosed as composite pheochromocytomaganglioneuromas. The proportions of ganglioneuroma in cases 1, 2, 3, 4, 5, and 6 comprised from 30%, 40%, 10%, 30%, 50%, and 60%, respectively.
DISCUSSION
Preoperative detection of a composite pheochromocytoma-ganglioneuroma is difficult because the low incidence of ganglioneuroma and the symptoms are not different from those of typical pheochromocytomas. Pathologically, the chromaffin cells of the pheochromocytoma are more readily highlighted with chromogranin and synaptophysin staining and they are admixed with S-100 protein-positive sustentacular cells [7]. All the tumor cells of the pheochromocytoma portion out of the composite tumor are strongly positive for tyrosine hydroxylase, i.e., the rate-limiting enzyme in catecholamine synthesis, while the ganglioneuroma portion displays neurofilament-positive mature ganglion cells and S-100 protein-positive Schwann cells.
Composite pheochromocytoma-ganglioneuromas are composed of chromaffin cells and the sympathetic ganglion cells [1-6]. The histogenesis of composite adrenal medullary tumors has been attributed to the common embryologic origin of the chromaffin and neuronal cells from the neural crest. To date, less than 50 cases of composite pheochromocytoma-ganglioneuromas have been reported since the first description [8]. The composite tumor has been used to describe tumors that theoretically arise from a common embryologic progenitor, whereas mixed pheochromocytomas designate tumors that have no common embryologic ancestry.
In about 70% of composite adrenal medullary tumors, the accompanying second tumor component was ganglioneuroma. The clinical symptoms associated with composite pheochromocytoma-ganglioneuroma are usually similar to those of ordinary pheochromocytoma, but secretion of vasoactive intestinal polypeptides (VIP) is more common with composite pheochromocytoma-ganglioneuroma compared to the latter [3]. Hypertension and its related symptoms are common in composite pheochromocytoma-ganglioneuroma. Occasional normotensive patients with composite tumors may be explained by the theory of autonomic regulation by ganglioneuroma, and it partially depends on the complex biochemical interaction and proportion of each element [9]. Rarely, watery diarrhea, hypokalemia and achlorhydria syndrome due to a functional VIP-secreting tumor can be observed in patients with composite pheochromocytoma-ganglioneuroma, and vasodilatory action of VIP also contributes the normotension [10]. Commonly observed muscle weakness may be related to associated hypokalemia [11]. Adrenal composite tumor may be incidentally discovered, masquerading as acute relapsing pancreatitis [5]. These composite tumors of the adrenal medulla may be associated with sporadic tumors or as part of neurofibromatosis type I, von Hippel-Lindau disease, MEN 2A and MEN 2B syndromes [12]. The present cases were shown to have no relation to neurofibromatosis, but 6% to 11% of composite pheochromocytomas are associated with genetic conditions. In general, the patients with pheochromocytoma are likely to develop papillary carcinoma, which may be related to the undulatory release of thyroid hormone secondary to circulating catecholamines [6,13]. Albeit rare, these findings should warn the patients to undergo regular examination of the thyroid gland.
Both pheochromocytoma and ganglioneuroma commonly manifests radiologically as a well-defined, smooth or lobulated mass with or without calcification. CT is accurate in the detection of pheochromocytomas. However, nearly one third of all cases show a non-specific appearance that may overlap with the appearance of adrenocortical carcinoma [14]. In the same manner, composite pheochromocytoma and ganglioneuroma shows heterogenous radiologic, gross and microscopic features with varying admixtures of ganglioneuroma and pheochromocytoma components [15].
The clinical significance of microscopically detecting the other component from pheochromocytoma has not yet been clarified because of the paucity of reports. Although there was one case of pheochromocytoma-ganglioneuroma showing hepatic metastasis, the composite pheochromocytomas with only a ganglioneuroma portion behave in the same manner as a classic pheochromocytoma and even when there are neuroblastomatous components because neuroblastomatous elements have low mitotic-karyorrhectic index and absence of N-myc amplification [15-17]. A review by Lam and Lo [15] suggests that the composite tumors are seen in older patients and are bigger than typical pheochromocytomas, which is possible that they are more aggressive than pheochromocytoma alone [10].
Here, all the six cases of composite pheochromocytoma-ganglioneuroma undertook laparoscopic adrenalectomies. Laparoscopic adrenalectomy provides the benefits of a minimally invasive approach, and is regarded to be the surgery of choice for pheochromocytoma, compared to open resection, due to the improved preoperative hemodynamic stability and the lessened need for postoperative analgesics [18].


 XML Download
XML Download