

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Neurofibromatosis type 1 (NF1) is one of the most common autosomal dominant inherited disorder affecting the nervous system and is associated with mutations in the NF1 gene. The cardinal features of NF1 are multiple dermal neurofibromas, café-au-lait spots, axillary or inguinal freckling, and hamartomas of the iris (Lisch nodules). NF1 is diagnosed by clinical criteria initially established by the National Institutes of Health Consensus Development Conference in 1988 and the criteria was revised by Gutmann et al. [2] in 1997. The NF1 gene is located on chromosome sub-band 17q11.2 and contains 57 exons spanning approximately 300 kb of genomic DNA (Fig. 1), and is transcribed to an mRNA of 11-13 kb that encodes for a protein, neurofibromin, of 2818 amino acids [3]. NF1 is caused by a loss of function of the NF1 gene, a tumor suppressor gene, which encodes neurofibromin, a GTPase activating protein involved in the negative regulation of Ras activity. Therefore, a neurofibromin deficiency results in hyperactivation of Ras, which is associated with cell proliferation and differentiation. GTPase activating protein-related domain (GRD), which is encoded by exons 20-27a, is one of the most important functional domains in neurofibromin. The interaction of Ras-GRD stimulates GTP hydrolysis which can mediate the regulation of cell proliferation and apoptosis. However, there are few studies about C-terminal region of NF1 gene and it has also yet to be known about the function of C-terminal area of NF1 gene.
In Korea, Jeong et al. [7] reported the spectrum of NF1 mutations in Korean NF1 patients, and discussed a genotype-phenotype correlation analysis in 2006. In the report, there was no detected mutation in C-terminal region, but most of mutations were found in GRD and cAMP protein kinase recognition sites (cAMP/PK).
We recently experienced two families of NF1 accompanied by pheochromocytoma and the mutations of two families have been found in C-terminal region of NF1 gene. Therefore, we performed the genetic analysis of the NF1 gene to verify the genotype-phenotype correlation in NF1 associated with pheochromocytoma.
CASE REPORT
1. Patients
1) Family 1
(1) Proband
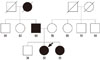
A 31-year-old woman suffered from intermittent paroxysmal headaches and chest discomfort. However, there were no abnormalities on the cardiologic evaluations and no improvement in the symptoms, despite continuous medication for 1 year. Finally, the patient was admitted to the hospital for evaluation of secondary hypertension. Based on history and clinical examination, the patient's sister and father were diagnosed with NF1 (Fig. 2). The physical examination revealed café-au-lait spots on the back and lower extremities and axillary freckling. We diagnosed her as NF1 by the clinical criteria.
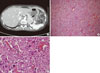
We obtained a 24-hour urine test for ruling out secondary hypertension, and the results revealed the following: vanillylmandelic acid (VMA), 30.8 mg/day (normal, 2-10 mg/day); and metanephrine, 3.94 mg/day (normal, 0.2-1.2 mg/day). Serum calcium, intact PTH and calcitonin were 10.2 (normal, 8.2-10.8 mg/dL), 24.39 pg/mL (normal, 10-65 pg/mL) and 4.91 pg/mL (normal, 0-10 pg/mL), respectively. Moreover, we confirmed a 4.0-cm well-enhancing mass in the left adrenal gland by abdominal computed tomography scan (Fig. 3A). Based on the results, we diagnosed the patient of a pheochromocytoma accompanied by NF1.
The patient underwent a left adrenalectomy and was evaluated for pheochromocytoma-associated syndrome with thyroid sonography, fundoscopic exam, brain magnetic resonance imaging (MRI), positron emission tomography, and a parathyroid hormone level. The results of almost all tests were negative, but the brain MRI revealed multiple subtle high signals on T2-weighted images and low signals on T1-weighted images in the left temporal lobe. The lesion was suspected to be a dysembryoplastic neuroepithelial tumor based on the imaging studies. As the location of the tumor was difficult to approach surgically, the patient has only been observed periodically.
(2) Sister
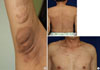
A 29-year-old woman was diagnosed with NF1 in 15 years ago. The patient had suffered from multiple plexiform neurofibromas involving both axillae (Fig. 4A). The neurofibromas continued to increase in number and size throughout adulthood, causing stinging, itching, pain, and disfiguration. Unlike her sister, she did not have hypertension or a pheochromocytoma. There were no other ocular or systemic diseases associated with NF1. The patient underwent surgery for disfigurated and symptomatic neurofibroma.
2) Family 2
(1) Proband
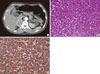
A 48-year-old man visited a clinic with paroxysmal headache, palpitation and blurred vision during 3 days. Systolic blood pressure was over 200 mmHg but his symptoms were not improved with antihypertensive medication. He thereby was evaluated by abdominal computed tomography (CT) for secondary hypertension, and it revealed a heterogeneous enhanced 5.3 cm sized mass in right adrenal (Fig. 5A). Therefore, he was referred to our hospital. He showed multiple café-au-lait spots, multiple colored papules and nodules on whole body which were seen at the age of 10 (Fig. 4B, C). Blood pressure was controlled by doxazocin initially, after then, by phenoxybenzamine 30 mg/day and maintained below 130/80 mmHg. Twenty-four-hour urine collection and analysis was done; VMA, metanephrine, epinephrine and norepinephrine were 12.23 mg/day (2-10 mg/day), 0.3 mg/day (0.14-0.78 mg/day), 51.95 µg/day (normal, 0-40 µg/day) and 172.72 µg/day (normal, 0-80 µg/day) respectively. Serum calcium level, intact PTH and calcitonin were normal at 9.3 mg/dL, 30.43 pg/mL, and 4.42 pg/mL. His skin biopsy result was dermal spindle cells which were positive for S-100 protein and negative for CD68, suggestive of neurofibroma. We diagnosed the proband with NF1 accompanied by pheochromocytoma. He underwent a right adrenalectomy and was evaluated for pheochromocytoma-associated syndrome with ophthalmologic examination, brain MRI and parathyroid level. There were all negative results in the examinations. Parents of the proband passed away a long time ago and he has no family, so genetic analysis of the family members was not performed.
2. Methods
The following gene analyses and fibroblast cultures were performed with written informed consent to confirm whether or not the patients or their family members had germline mutations. The study was approved by Institutional Review Board of Gachon University Gil Hospital, Korea.
3. Comprehensive NF1 mutation analysis
Comprehensive NF1 mutation analysis starts with extraction of DNA from the patient's blood sample. Another aliquot of the blood sample is used to start a short-term culture of phytohaemagglutinin-stimulated lymphocytes. Total RNA is extracted from puromycin-treated cultures and cDNA is prepared.
Four intragenic microsatellite polymorphisms, i.e., IVS27 CAGT, AAAT-Alu IVS27, IVS27 GT, and IVS38 TG53.0, and 1 extragenic marker, i.e., 3'NF1-1, are analyzed as a first approach to determine whether 2 copies of the NF1 gene are likely to be present. If no heterozygous signal is observed for any of these markers, FISH analysis is performed using PACs 926B9 and 1002G3 to confirm or rule out the presence of a total gene deletion.
Thereafter, the total coding region of the NF1 exon is analyzed by long-range RT-PCR and direct cycle sequencing, starting from 3 overlapping RT-PCR fragments, spanning exons 1-35b, 20b-42, and 30-57. Sequencing is performed using dye-terminator chemistry on an ABI PRISM 3730xl capillary sequencer; the NF1 exon 1 is hereafter sequenced at the gDNA level.
If no mutation is identified by direct cycle sequencing, a multiple-ligation-probe assay (MLPA) is performed to detect the copy number changes (deletions/duplications) in the coding region of the NF1 gene. A routine cytogenetic analysis is not performed.
4. Pathologic findings of the tumor
1) Family 1
(1) Proband
Gross examination of the adrenal gland was presented with a yellow cortical lesion and a central hemorrhagic area. The size of the tumor was approximately 2.5 × 2.0 cm in size. The tumor consisted of small nests and alveoli patterns, which are the one of the typical manifestation of pheochromocytoma. Cellular and nuclear pleomorphism was often presented in the specimen. Giant and bizarre cells were also commonly observed; however, mitotic figures were rare (Fig. 3B, C). The tumor cells stained positive for synaptophysin and chromogranin, which are characteristic features for pheochromocytoma.
(2) Sister
Histopathologic findings of the tumor are consistent with plexiform intraneural neurofibroma showing biphasic pattern; one is perineural patterns which are composed of loosely arranged bipolar and tripolar cells in a myxoid matrix and the other is schwannian patterns showing thin spindle cells with elongated, wavy nuclei associated with wavy collagen bundles.
2) Family 2
(1) Proband
Gross examination of the adrenal gland had 6.0 × 5.0 × 2.2 cm sized, yellowish brown colored and well-demarcated partially cystic mass. The tumor cells stained positive for S-100 protein and chromogranin. Pathologic finding of this tumor was compatible with pheochromocytoma with no necrosis, mitosis and capsular invasion (Fig. 5B, C).
5. NF1 mutation producing splicing defects and affecting truncated protein
1) Family 1
The patient and her family members exhibited mutations involving the splice site in lymphocytes and adrenal tissues (Fig. 6A). No other possible damaging alteration was detected after comprehensive NF1 mutation analysis. This finding confirmed the diagnosis of NF1 in this patient. The detected point mutation (c.7907+1G>A) caused skipping of exon 53 (Fig. 6B). In the process of translation, the mutation, c.7907+1G>A, made a terminal codon (TAA) during mRNA splicing and 53 exon skipping caused premature termination of protein translation.
2) Family 2
The point mutation (c.5206-8C>G) was detected and that is out of frame for splicing of exon 37 caused by mutation of NF1 gene (Fig. 7A). This alteration creates a novel splice acceptor that is used in lieu of the native splice acceptor, and an out of frame insertion of intronic sequence into the NF1 mRNA occurs (Fig. 7B).
DISCUSSION
NF1 is a common autosomal dominant genetic disorder caused by mutations in the NF1 gene. Pheochromocytomas consist of minor, but established features of NF1. Because of the low prevalence of pheochromocytomas reported in patients with NF1, it has remained the question of whether or not pheochromocytomas are a true component of NF1, or a coincidental finding. However, recent studies have shown that loss of the remaining wild-type allele is observed in NF1-related pheochromocytomas.
There is no clear relationship between specific NF1 mutations and clinical features. The proband and her sister showed the same genomic mutation for the NF1 gene, but each of them had a different phenotype. They shared the clinical features of café-au-lait spots and axillary freckling, but the proband had a pheochromocytoma and dysembryoplastic neuroepithelial tumors, while her sister had only multiple neurofibromas. This suggests that NF1 allelic heterogeneity may not affect phenotypic differences. Genotype-phenotype correlation has not been recognized because of the complexity of the NF1 phenotype and the heterogeneity of pathogenic NF1 mutations.
We detected the point mutations (c.7907+1G>A and c.5206-8C>G) which caused frame shifting of exon 53 and 37, respectively. These mutations are classified as the intronic nucleotide change of type I mutations by classification of NF1 mutations affecting splicing according to the five group classes. Pros et al. [14] detected 282 different mutations in the NF1 gene and analyzed the mRNA effect. In his report, there was a similar mutation (c.7907+6T>C), which caused the same result of family 1 as skipping of exon 53 on mRNA splicing.
GTPase activating protein (GAP) is important in the negative regulation of Ras activity. GAP has been recognized to interact with the Ras protein to stimulate hydrolysis of GTP to turn off the Ras oncogene protein. The GAP-related domain, which is encoded by exons 20-27a, is one of the most important functional domains in neurofibromin. Many mutations in the GAP-related domain of the NF1 gene were identified to interfere with Ras signaling pathways and to cause to the development of tumors. Some studies verified that restoration of the NF1-GAP-related domain was able to reduce Ras activation in neurofibromin-deficient Schwann cells and caused a decline in the proliferation of Schwann cells from both neurofibromas and malignant peripheral nerve sheath tumors.
The GAP-related domain of neurofibromin is not the only domain underlying disease manifestations. In fact, at least a one-fourth of NF1 mutations are in the GAP-related domain. However, Bausch et al. [16] reported that 35% of the germline mutations in NF1-related pheochromocytomas are within the cysteine-serine-rich domain other than GAP-related domain. This result reveals that the cysteine-serine-rich domain is also an important functional domain and plays a prominent role in the formation of NF1-related pheochromocytomas.
Neurofibromin can confer sensitivity to apoptosis and regulate cell proliferation with Ras-independent pathways. Molecular and genetic studies in mammals and Drosophila have revealed that neurofibromin inhibits Ras activity and regulates adenylyl cyclase activity and cAMP levels. The Ras-GAP interaction of NF1 is necessary for Ras/NF1-dependent adenylyl cyclase signaling, but not NF1/Gαs-dependent adenylyl cyclase signaling. The latter pathway is necessary for part of the C-terminal residues downstream of the GAP-related domain. In fact, reconstitution of the NF1-GAP-related domain can result in a partial restoration of cAMP levels in NF1 -/- astrocytes. Therefore, we suggest that another region of the NF1 gene, rather than the GAP-related domain is associated with formation of the tumor or signal transduction pathway which can control angiogenic factor expression.
The suitability of using antisense morpholino oligomers (AMO) to restore the aberrant splicing caused by deep intronic mutations in the NF1 gene has been reported. The authors showed corrected aberrant splicing and increased levels of wild-type NF1 transcripts with AMO treatment. Moreover, they revealed that correction of aberrant NF1 splicing decreased the active Ras-GTP levels in primary fibroblasts, which is consistent with the restoration of neurofibromin GTPase activity to wild-type levels.
We detected germline NF1 mutations in patients with a pheochromocytoma displaying the NF1 phenotype; they are novel mutations which are thought to be related to pheochromocytomas. Further studies are needed to clarify the genotype-phenotype correlations of pheochromocytoma occurred as a component of NF1.
1. Summary
NF1 is one of the most common autosomal dominant inherited disorder affecting the nervous system and is associated with mutations in the NF1 gene. Pheochromocytomas consist of minor, but established features of NF1. We recently experienced two families of NF1 accompanied by pheochromocytoma and detected novel germline mutations in the patients. Two novel mutations have been found in C-terminal region of NF1 gene contrast to the former reports. Therefore, we should also importantly consider the function of C-terminal region of NF1 gene in NF1-related pheochromocytoma. Further studies are needed to clarify the genotype-phenotype correlations of pheochromocytoma occurred as a component of NF1.



 XML Download
XML Download