

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Multiple endocrine neoplasia (MEN) type 2A is an autosomal dominant condition characterized by the development of medullary thyroid cancer (MTC), pheochromocytoma, and parathyroid hyperplasia. In 1993, Mulligan et al. [1] reported that MEN 2A could be traced to a germline mutation in the RET gene. The development of pheochromocytoma in 80% of patients with MEN 2A is due to mutations in codon 634 [2]. Pheochromocytoma is usually diagnosed at the same time as MTC and is the first sign of MEN 2A in 9-27% of cases [3]. Between 30% and 50% of pheochromocytomas in MEN 2A patients are asymptomatic [3,4], given that the tumors are diagnosed by screening. However, pheochromocytoma presenting as fatal cardiogenic shock is a recognized and relatively uncommon occurrence, with only a few cases reported [5].
In this rare case, we diagnosed pheochromocytoma presenting with symptoms of acute heart failure and pulmonary edema due to catecholamine-induced cardiomyopathy. We subsequently diagnosed MTC in the patient's thyroid nodules. We confirmed that the patient had a mutation of C634R in exon 11. Based on our findings, the patient's family members underwent screening tests and RET gene tests for MEN 2A. In a younger sister of the patient, who was undergoing follow-up observations for benign thyroid nodules at a private clinic, the same mutation was noted, and MTC was also identified. We report our findings regarding this family and provide a review of the relevant literature.
CASE REPORT
We report the case of a 29-year-old woman presenting with sudden respiratory distress and chest pain. According to her past history, the patient had undergone a normal delivery five months prior and had no previous medical history noted at the time of the prenatal exam. She had received a medical examination at a medical center two months before the incident, and all results fell within normal limits, including blood pressure, electrocardiogram (ECG), chest X-ray and thyroid function tests. The patient had experienced frequent episodes of fever, sweating, and fatigue in the month prior to presentation. She had visited a private clinic, where her blood pressure was determined to be normal and no evidence of goiter was found. However, her physician prescribed oral beta-blockers (propranolol 40 mg) and propylthiouracil (PTU 50 mg) suspecting hyperthyroidism or postpartum thyroiditis.
Upon admission, the patient exhibited dyspnea and chest pain. Initially, heart enzymes were elevated and an ECG showed a depression in the ST-segment from V4 to V6 in the inferior walls and akinetic wall motion in the lateral walls in echocardiography. She developed radiologic and echocardiographic findings of acute pulmonary edema and heart failure, possibly caused by ischemic heart disease. Likewise, her blood pressure could not be ascertained and her pulse was weak. She underwent endotracheal intubation and was treated using a mechanical ventilator, intravenous nitroglycerine, heparin, and dopamine. Coronary angiography was not performed due to the clinical course aggravation. After conservative management, the patient's pulmonary edema and heart failure improved. On the third day, she developed persistent oliguria due to hypovolemic shock, and an increase in BUN/creatinine up to 78/9.1 mg/dL was noted. There were no specific lesions found upon abdominal ultrasonography. Thereafter, the patient was transferred to our hospital for further evaluation and treatment. After the patient's symptoms improved, with the exception of the oliguria, 24-hour ECG monitoring was performed in order to investigate the possibility of ischemic heart disease as a cause of acute pulmonary edema; however, no abnormal findings were observed. In addition, echocardiography and a 99-m thallium scan also showed no findings suggestive of ischemic heart disease, and serological tests to identify etiologic factors for acute heart failure produced no abnormal findings. Physical examination of the patient's head and neck region revealed a movable, non-tender nodule in the thyroid gland without goiter. Thyroid function tests, TSH-binding inhibitory immunoglobulin (TBII), anti-TPOAb, and anti-TgAb were all within normal ranges. Ultrasonography and thyroid scans revealed two cold nodules sized 1.7 cm on the right and 1.9 cm on the left. Fine needle aspiration (FNA) of these nodules demonstrated the presence of benign follicular cells. The patient underwent hemodialysis in our hospital, and her renal function and symptoms ameliorated. She was subsequently discharged.
Two weeks following discharge, the patient complained of palpitations, headache, nausea, and vomiting and was readmitted for further evaluation. Upon physical examination, her vital signs were blood pressure, 170/110 mmHg; pulse, 100/min; breaths, 20/min; temperature, 36.7℃. Physical examination of the patient's head and neck region revealed a movable nodule in the thyroid gland without lymphadenopathy. There were no abnormal findings in the chest and abdomen. Her complete blood count did not show any abnormal findings. The patient's BUN/creatinine had increased to 29/2.4 mg/dL, but her liver function was within the normal range. Accordingly, we began to suspect and investigate the possibility of pheochromocytoma. Plasma epinephrine was 2.54 ng/mL (normal range 0.07-0.4), and norepinephrine was 1.37 ng/mL (normal range 0.04-0.2). Based on the 24-hour urine collection analysis, vanillylmandelic acid (VMA) was 10.7 mg/day (normal range 1-5), metanephrine was 2.9 mg/day (normal range 0.2-1.2), epinephrine was 68.0 µg/day (normal range 15-60), and norepinephrine was 87.0 µg/day (normal range 1.2-20). An abdominal MRI was performed to identify the location of the tumor, and it indicated a mass approximately 3 cm in diameter in the left adrenal gland (Fig. 1A). 131I-MIBG scans (Fig. 1B) revealed an increased isotope uptake in the left adrenal gland, and thus, a left adrenalectomy was performed. A brown mass 2.9 × 3.3 × 4.9 cm in size was identified as the cause of pheochromocytoma based on histopathology (Fig. 2). Following the adrenalectomy, 24-hour urine catecholamine and metabolites decreased to normal ranges. In order to evaluate the possibility of MEN2A, based on the histopathological confirmation of pheochromocytoma and palpation of the thyroid nodule, serum calcitonin level was measured and found to be elevated to 1,306 pg/mL (normal value < 170), whereas the serum calcium and intact PTH levels were within normal limits. The increased level of serum calcitonin led us to suspect the presence of MTC, and thus, under the suspicion of MTC in MEN 2A, a total thyroidectomy and lymph node dissection were performed. An oval-shaped solid mass was detected in each of the thyroid lobes, and histopathological examination confirmed the diagnosis of MTC (Fig. 3). Normal parathyroid gland tissue was also detected during the surgery and was preserved.
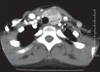
Based on our findings, the patient was diagnosed with MEN 2A, and her family members underwent screening tests and RET gene tests for MEN 2A. During familial screening, the patient's 27-year-old younger sister was found to have been diagnosed with benign thyroid nodules three years prior. We evaluated the thyroid nodules from the patient's sister. She had no particular past medical history, and her physical examination and laboratory findings did not produce any particular findings other than those in the head and neck region, which revealed a movable and non-tender nodule without lymph node enlargement. Thyroid function and autoantibody tests were all normal. Likewise, all laboratory data related to pheochromocytoma were within normal ranges. However, the patient's serum calcitonin level was increased to 1,303 pg/mL (measured without stimulation), whereas intact PTH and calcium levels were within normal limits. A subsequent neck CT scan (Fig. 4) revealed bilateral thyroid nodules in multiple sites. The patient underwent a total thyroidectomy and total lymph node dissection. An oval-shaped solid mass was detected in each of the thyroid lobes, and histopathological examination confirmed the diagnosis of MTC (Fig. 5). The presence of normal parathyroid glands was confirmed during surgery.
With the consent of the patient and family members, we performed genetic studies to determine whether or not germline mutations of RET genes were responsible for MEN 2A. Genomic DNA was extracted from peripheral blood using the Wizard Genomic DNA Purification Kit (Takara, Japan). Automatic nucleotide sequence analysis was performed for exons 10, 11, 13, and 14. In the patient, we identified a point mutation where TGC (cysteine) was replaced by CGC (arginine) at codon 634 in exon 11 of the RET gene; the patient's younger sister had the same mutation. Genetic testing of the patient's family members, including her mother, brother, and another sister, did not reveal any abnormal findings suggestive of genetic mutations (Fig. 6).
Discussion
MEN is an autosomal dominant disease in which various endocrine tumors develop secondary to a genetic mutation. Based on the clinical manifestations, MEN can be classified as Type 1, 2A, 2B, or other. Type 2A accounts for 75% of all cases of MEN and is characterized by MTC, pheochromocytoma, and parathyroid hyperplasia or adenoma [6]. Pheochromocytoma manifests a broad spectrum of clinical symptoms and signs, ranging from asymptomatic presentation to paroxysmal hypertension, sweating, and palpitations due to excitation of sympathetic nerves [7]. However, as was the case for the patient in our study, the clinical symptoms of heart failure accompanied by acute pulmonary edema are not commonly observed [7]. Pheochromocytoma should also be included in the differential diagnosis of acute coronary syndrome, because acute catecholamine secretion may induce chest pain and abnormal ECG changes mimicking acute myocardial infarction, especially in young patients with low cardiovascular risk [8]. Tumor necrosis caused by ischemia secondary to vasoconstriction and a massive release of catecholamines also contribute to initial hypertension as well as subsequent hypotension and myocardial damage [9]. Unusual findings of ST-segment depression on EKG and other findings on echocardiography can be misinterpreted as an acute ischemic heart disease due to atherosclerotic coronary artery disease. Likewise, cardiac diseases caused by pheochromocytoma are not easily diagnosed because the high dose of catecholamines released from a pheochromocytoma promotes myocardial metabolism and induces the constriction of coronary arteries [8]. Most MEN patients have no significant coronary atherosclerosis, in contrast to cases of classic myocardial infarction [10]. In addition, acute renal failure may be caused by intense renal vasoconstriction induced by catecholamines and cardiovascular shock [11]. In our case, we inferred that an acute spontaneous hemorrhage within the tumor mass led to a massive release of catecholamines (as demonstrated by pathological findings), which resulted in acute heart failure and pulmonary edema that led to subsequent cardiogenic shock and acute renal failure due to ischemic injury. Pheochromocytomas in MEN 2 patients do not secrete catecholamines as readily or as continuously, and have a high capacity to synthesize hormones, especially adrenaline [12,13]. So, the major symptom is more paroxysmal than chronic, and other minor symptoms are less frequent than in the sporadic cases. Fortunately enough, our presented case did not have any of the typical symptoms at delivery. However, the administration of beta-blockers (propranolol) for symptoms of palpitations, headaches, and fatigue may have aggravated the patient's heart failure caused by catecholamine-induced cardiomyopathy. Catecholamine-induced cardiomyopathy in patients with a pheochromocytoma appears to be completely reversible and potentially curable if timely adequate treatment occurs; otherwise, the resulting acute crisis may be fatal [8].
A genotype-phenotype relationship has been demonstrated between RET mutations and pheochromocytoma. A review of cases reported in Korea [14-16] showed that the mean age of patients with MEN 2A is 38 years, and variations in codon 618 are seen in 20% of exon 10 segments, while the remaining 80% are coding variations in exon 11. Of these variations, the frequency of C634R is the highest (53%). In particular, mutations of codon 634 in exon 11, which appear with the highest frequency, are closely associated with pheochromocytoma and hyperparathyroidism [17]. In our patient's family, a germline mutation causing a cysteine to arginine conversion at codon 634 of exon 11 of the RET gene was identified. Accordingly, in the younger sister of the patient, tests for pheochromocytoma and follow-up observations based on serum concentrations of parathyroid hormone and calcium performed on a yearly basis are warranted. It has also been reported that mutations of codon 634 are associated with the development of pheochromocytoma before the age of 5-10 years [18]. In addition, this mutation corresponds to the high-risk phase II group with respect to the degree of malignancy of MTC [6]. Thus, RET genetic testing and meticulous follow-up observation will be necessary for the patient's daughter.
At the present time, FNA is the standard diagnostic test for identification of thyroid nodules, and the accuracy of FNA tests for thyroid cancer is 90% [19]. In cases of papillary cancer, the accuracy of FNA diagnosis reaches approximately 93%, but it is only 79% with respect to correct diagnosis of medullary cancer [19]. In this case, the patient's thyroid nodule had been identified as a benign follicular nodule on FNA, but increased levels of serum calcitonin were noted. Thus, we suspected medullary thyroid carcinoma, the diagnosis of which was confirmed at surgery. Therefore, when managing a patient with a thyroid nodule, a thorough inspection of family history is important. In cases where MTC is clinically suspected, meticulous laboratory studies including serum calcitonin levels is necessary.
SUMMARY
We suspected the presence of pheochromocytoma and were able to confirm our diagnosis in a patient presenting with symptoms of fatal cardiogenic shock due to catecholamine-induced cardiomyopathy. Furthermore, we evaluated the possibility of MEN 2A as the source of the thyroid nodules and diagnosed MTC using various tests and surgery. The patient had a younger sister who underwent follow-up observation for benign thyroid nodules, and as a result, the diagnosis of MTC was established during selective testing for MEN 2A. Genetic testing for the RET gene demonstrated that the diagnosis of MEN 2A was due to a mutation of C634R on exon 11. Using a selective test for familial genes, mutations of the same genes were confirmed, and a family of MEN 2A individuals was identified.





 XML Download
XML Download