

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Obesity has become a huge burden worldwide. According to the 2013 Korea National Health and Nutrition Examination Survey, one in three Korean adults are obese and more than half are considered to be at least overweight [1], implying that Korea is not an exception to this worldwide pandemic. Environmental factors such as unhealthy dietary habits, sedentary lifestyle and socioeconomic influences are considered as the major contributors.
In the past several decades, evidence suggests that obesity is a state of chronic, low-grade inflammation. One of the earliest evidence demonstrated the expression of proinflammatory cytokine, tumor necrosis factor α (TNF-α), in rodent adipose tissue during the development of obesity and the attenuation of insulin resistance followed by neutralization of TNF-α [2]. This study supports the role of inflammation in obesity and regulation of its complications by inflammatory mediators [3]. Thereafter, significant advances in understanding the complex role of immune-metabolism have been accomplished, and obesity is now known to be associated with proinflammatory cytokine secretion, immune cell infiltration, and disrupted function of tissues involved in glucose homeostasis, leading to insulin resistance [4].
Nitric oxide (NO) is an endogenous signaling molecule produced by nitric oxide synthase (NOS). NO is involved in various physiologic processes such as the regulation of synaptic transmission, vasodilation, leukocyte-endothelial interactions, immune function, and angiogenesis [5]. Besides these pleiotropic effects, NO has recently emerged as an important regulator of energy metabolism. For example, NO bioavailability reflected by NOS activity is decreased in animal models of diet-induced obesity (DIO) [6] and in obese and insulin-resistant patients [78]. The reduction of NO content occurs early in the course of developing insulin resistance, followed by vascular and peripheral tissue (such as liver, muscle, and adipose) inflammation and insulin resistance during dietary excess [910]. Furthermore, genetic deletion of endothelial nitric oxide synthase (eNOS) is associated with insulin resistance even in the absence of high-fat (HF) stimuli [11]. Conversely, interventions to increase NO output has remarkable effects on obesity and insulin resistance in both animal and human studies [121314], implying that anti-inflammatory and anti-obesity properties of NO might serve as a potential therapeutic approach against obesity and its related metabolic outcomes.
Vasodilatory-stimulated phosphoprotein (VASP) is a 40 kDa protein associated with cell adhesion and modulation of cytoskeletons [1415] and found in various cell types, but is highly expressed in vascular endothelial cells, smooth muscle cells, fibroblasts, and platelets [15]. VASP is also known to be a downstream molecule of NO signal transduction pathway. Recently, a growing body of evidence suggests that VASP as a downstream mediator of NO signal transduction pathway exerts anti-obesity effects by improving inflammation and insulin resistance in various tissues involved in glucose metabolism [1011161718]. Therefore, in this review, we evaluate the current evidence supporting the protective effect of NO/cyclic guanosine monophosphate (cGMP)/VASP signaling pathway against obesity and possibly, type 2 diabetes mellitus.
Go to : 
NO/cGMP/VASP SIGNALING PATHWAY
The eNOS is mainly expressed in endothelial cells and generates eNOS-derived NO [19]. In cardiovascular tissues, eNOS-derived NO is known to exert vasodilatory, anti-inflammatory, and anti-proliferative effects [1920] via cGMP-dependent protein kinase (PKG) by increasing cGMP levels [9]. The NO/cGMP-induced PKG activation in various cell types exerts diverse biological function by modulating its multiple downstream effectors. For example, PKG interacts with VASP [21222324] and survival molecules such as the apoptosis-regulating protein BAD [25], the oncogenic tyrosine kinase c-Src [22], and the transcription factor cyclic adenosine monophosphate (cAMP)-responsive element binding protein (CREB) [26] to regulate cell migration, survival, and proliferation. On the other hand, PKG-mediated activation of the mitogen-activated protein kinase (MAPK) family regulates angiogenesis in vascular endothelial cells [27] and the contractile function of myocardial fibers [28] and vascular smooth muscle cells (Fig. 1) [29].
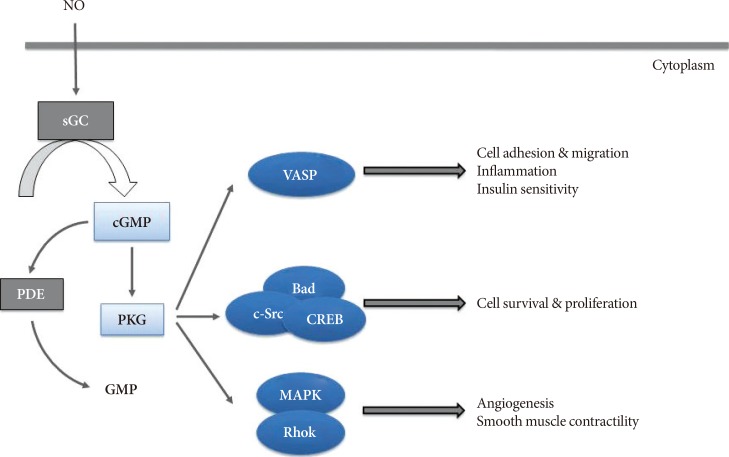 | Fig. 1Schematic illustration of nitric oxide (NO)/cyclic guanosine monophosphate (cGMP) signaling pathway and its downstream effects. Activated cGMP-dependent protein kinase (PKG) by NO/cGMP signaling exerts diverse biological function by modulating its multiple downstream effectors in various cell types. PKG interacts with vasodilatory-stimulated phosphoprotein (VASP) to regulate cell adhesion and migration, inflammation and insulin sensitivity, as well as with survival molecules to regulate cell survival, and proliferation. PKG also exerts its role in angiogenesis and smooth muscle contractility via mitogen-activated protein kinase (MAPK) family. sGC, soluble guanylyl cyclase; PDE, phosphodiesterase; c-Src, proto-oncogene tyrosine-protein kinase c-Src; CREB, cyclic adenosine monophosphate (cAMP)-responsive element binding protein; RhoK, rho-associated protein kinase.
|
Of numerous various downstream effectors of PKG, implication of modulating VASP in cancerous or neural cells has been widely studied due to its role in cell adhesion, migration, and proliferation [21222324]. However, recently, phosphorylation of VASP by PKG on a specific serine residue (Ser-239) has been found to improve inflammation and insulin resistance in various peripheral organs such as the adipose tissue, the liver, and the vasculature [1011161718].
Go to :
NO/VASP SIGNALING IN ADIPOSE TISSUE
In visceral adipose tissue, nutrient excess leads to the infiltration of macrophages via the production of monocyte chemoattractant protein 1 (MCP-1), a key mediator recruiting macrophage precursors into the adipose tissue [3031]. Subsequent release of proinflammatory cytokines such as interleukin 6 (IL-6) and TNF-α results in impaired insulin signaling [3]. Hence, improving obesity-induced insulin resistance by regulating inflammatory signaling has been of great interest as it can serve as a potential therapeutic target.
Based on the anti-inflammatory role of endothelial NO signaling in the vasculature and the decline of vascular NO levels early in the course of DIO, Handa et al. [10] made an attempt to unravel the role of NO/cGMP/VASP signaling pathway in the pathogenesis of obesity-associated inflammation in adipose tissue. In this study, HF feeding significantly reduced phospho-eNOS and phospho-VASP in white adipose tissue, compared to mice fed a low-fat (LF) diet. Genetic deficiency of eNOS induced increased expression of proinflammatory cytokines in adipose tissue of eNOS–/– mice; however, a LF diet could not prevent adipose tissue inflammation in these mice, further reflecting the significance of endothelial NO signaling in attenuating adipose tissue inflammation. By contrast, administration of sildenafil, a drug that increases signaling downstream of vascular NO, prevented HF diet-induced proinflammatory gene expression and adipose tissue macrophage infiltration while improving insulin sensitivity in wild type (WT) mice [10].
Because the authors hypothesized that VASP, a downstream target of the NO/cGMP pathway, is crucial to the anti-inflammatory effects of NO signaling pathway on adipose inflammation, they used VASP–/– and WT littermate mice fed an LF and HF diet [10]. As expected, in VASP–/– mice, levels of mRNA encoding proinflammatory cytokines MCP-1, IL-6, and TNF-α were markedly elevated compared with WT controls [10]. The recapitulation of the effect of eNOS deficiency to increase adipose tissue inflammation in VASP-null mice suggests that VASP must be present for NO/cGMP signaling pathway to exert its anti-inflammatory effect in adipose tissue.
Go to :
NO/VASP SIGNALING IN THE LIVER
The liver is a pivotal player in glucose metabolism and inflammatory responses [32]. It is highly vascularized receiving 20% of cardiac output, and hepatic sinusoidal endothelial cells make up half of nonparenchymal cells of the liver [33]. Sinusoidal endothelial cells have shown to express eNOS and produce NO basally, regulating hepatic vascular resistance [34]. In a DIO setting, reduced vascular NO levels were followed by enhanced liver nuclear factor-κB (NF-κB) signaling and impaired insulin-mediated phosphorylation of Akt [35], highlighting the possible role of NO signaling in regulation of hepatic insulin resistance and inflammation.
Macrophages are myeloid-derived mononuclear cells that are a critical component of the innate immune response. They are more enriched in the areas that are exposed to pathogens, toxins and tissue damages such as the lung and the liver. Accumulating evidence supports a role for tissue macrophages in a broad spectrum of inflammatory conditions [36], and the number and activity of macrophages are associated with insulin resistance and metabolic deterioration in states of over nutrition such as obesity [3738].
Kupffer cells, the liver-specific macrophages, reside on the luminal side of the sinusoidal endothelium, and are mainly involved in scavenging, host immunity and immune tolerance in the liver [39]. Recent studies demonstrated that increased Kupffer cell activity is responsible for hepatic insulin resistance [4041], and that depletion of Kupffer cells attenuated the development of hepatic steatosis and hepatic insulin resistance, both of which suggest an important early role of Kupffer cells in diet-induced alterations in hepatic insulin resistance [19].
Tateya et al. [11] questioned whether endothelial NO signaling contributes to the effect of HF feeding to induce inflammatory activation of Kupffer cells and associated hepatic insulin resistance. In the study, proinflammatory activation of Kupffer cells was induced both in WT mice fed HF diet and in genetically eNOS-deficient mice, highlighting the protective effect of NO signaling in hepatic inflammation and insulin resistance [11]. Similarly, targeted deletion of VASP in vivo, a key downstream target of endothelially derived NO, predisposed to hepatic and Kupffer cell inflammation regardless of diet, whereas agent-enhanced VASP signaling reversed the inflammatory activity and insulin resistance in VASP-deficient hepatocytes and macrophages in vitro [11]. Subsequently, Tateya et al. [18] have also demonstrated that NO/VASP signaling increases hepatic fatty acid oxidation by activating AMPK in mice models, suggesting that direct activation of VASP could be a potential therapeutic target of hepatic steatosis.
The mechanism through which endothelial NO/cGMP/VASP signaling exerts protective effect against inflammation and insulin resistance in Kupffer cells has also been explored [17]. Using mice model with transgenic eNOS overexpression, this study demonstrated that the protective effect of NO signaling against HF diet-induced hepatic inflammation and insulin resistance is associated with reduced proinflammatory M1 and increased anti-inflammatory M2 activation of Kupffer cells [17]. Similar effects were induced by overexpression of VASP in macrophages, whereas VASP deficiency induced proinflammatory M1 macrophage activation. In attempt to determine whether VASP deficiency, specifically in the macrophage compartment, is sufficient to explain the hepatic inflammation and hepatic insulin resistance, transplantation of bone marrow from VASP-deficient donor mice into normal recipients was performed. As a result, the transplantation led to hepatic inflammation and insulin resistance resembling that induced in normal mice fed HF diet [17]. Taken together, NO/VASP signal transduction inhibits proinflammatory M1 activation of the Kupffer cells, and it can serve as a physiological determinant of macrophage polarization and a promising therapeutic target to prevent hepatic inflammation and insulin resistance.
Go to :
NO/VASP SIGNALING IN VASCULAR ENDOTHELIUM
Metabolic deterioration related to obesity is linked with cardiovascular diseases, and atherosclerosis is responsible for the vast majority of these cardiovascular events. Thus, it is important to detect and delay the progression of atherosclerosis in its early stage. In recent years, it has become evident that insulin resistance and endothelial dysfunction play a central role in the pathogenesis of atherosclerosis [42]. Metabolic dysfunction causes lipid deposition and oxidative stress to the vessel wall, triggering an inflammatory reaction, and the release of chemoattractants and cytokines worsens the insulin resistance and endothelial dysfunction [4243]. Within this context, therapies that improve vascular insulin resistance and inflammation would be ideal as it may reduce cardiovascular morbidity and mortality in clinical settings.
Reduced NO bioavailability occurs within 1 week of HF feeding in mice, causing endothelial cell to be significantly more vulnerable to the inflammatory effects of excess nutrition [35]. This further reduces NO production, leading to a "vicious cycle" of increased vascular inflammation and reduced NO levels [16]. By contrast, increasing downstream NO signaling in mice fed HF diet attenuates vascular inflammation [44]; thereby, breaking this vicious cycle and restoring vascular insulin sensitivity [16].
Cheng et al. [16] investigated the role of VASP as a downstream mediator of the anti-inflammatory effect of NO signaling in vascular tissue. Compared to mice fed a LF diet, markedly reduced levels of VASP Ser239 phosphorylation, a marker of VASP activation, were observed in aortic tissue of DIO mice. HF feeding was associated with increased aortic inflammation, as measured by increased NF-κB dependent gene expression, and reduced vascular insulin sensitivity (including insulin-stimulated phosphorylation of eNOS and Akt) [16]. These HF-diet-induced responses recapitulated in VASP–/– mice, whereas overexpression of VASP in endothelial cells blocked inflammation and insulin resistance induced by palmitate, reflecting the protective effect against inflammatory signaling and insulin resistance in vasculature [16]. These findings implicate that VASP can serve as a potential therapeutic target in the treatment of obesity-related vascular inflammation and insulin resistance.
Go to :
CONCLUSIONS
Peripheral insulin resistance in obese state is crucial to the etiology of metabolic disorders such as type 2 diabetes mellitus [45], and thus, understanding the molecular mechanisms of insulin resistance in the organs involved in glucose homeostasis can provide further insights to the development of potential targeted therapy. The series of studies we have evaluated in this review commonly indicate that NO/cGMP signaling limits obesity-related inflammation and insulin resistance in multiple organs, such as adipose tissue, liver, and vascular endothelium and also that VASP is a critical downstream mediator required for this protective effect of NO/cGMP signaling (Fig. 2). Developing potential drugs that alter NO signaling has been of caution due to its cytotoxic effect at high levels, generated by inducible NOS. However, targeting downstream molecules of NO-cGMP signaling pathway, such as soluble guanylyl cyclase or PKG could potentially overcome this drawback NO-targeted therapeutic approach possesses [10]. In this standpoint, VASP can be a promising therapeutic target to limit peripheral insulin resistance in obese state, although accumulation of more evidence is necessitated.
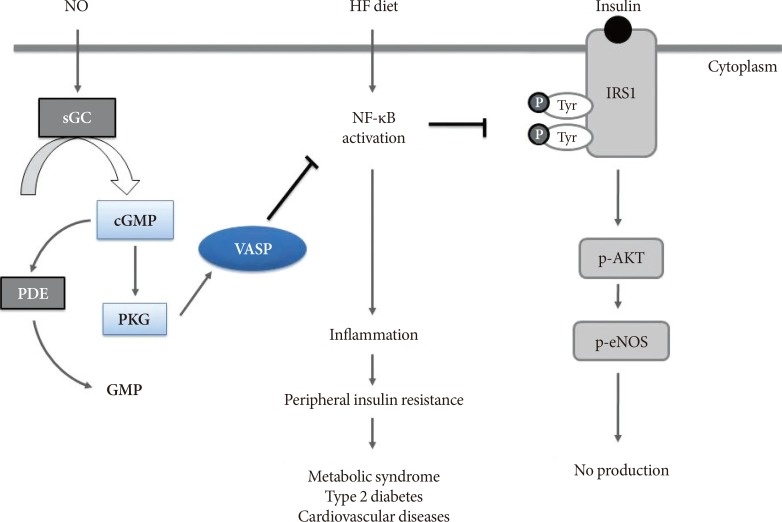 | Fig. 2Proposed effect of vasodilatory-stimulated phosphoprotein (VASP) on insulin resistance and inflammatory signaling in multiple organs. Nitric oxide (NO)/cyclic guanosine monophosphate (cGMP) pathway activates VASP, which inhibits nuclear factor-κB (NF-κB) activation; thereby, further inhibiting downstream inflammatory processes and enhancing insulin sensitivity. The schematic end-result of NO/cGMP/VASP signaling suggests its role against cardiometabolic disorders such as metabolic syndrome, type 2 diabetes mellitus, and cardiovascular diseases. Adapted from Cheng et al. [16], with permission from The American Physiological Society. HF, high-fat; sGC, soluble guanylyl cyclase; PDE, phosphodiesterase; PKG, cGMP-dependent protein kinase; IRS1, insulin receptor substrate 1; p-AKT, phospho-Akt; p-eNOS, phospho-endothelial nitric oxide synthase.
|
Go to :
 XML Download
XML Download