

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
The endoplasmic reticulum (ER) has diverse biological functions associated with cellular homeostasis and whole organism physiology. These include the biosynthesis, folding, posttranslational modification, assembly, and trafficking of membrane and secretory proteins [1]. Thus, the ER provides quality control of those proteins, which ensure that only properly folded proteins are exported via the secretory pathway. Unfolded or misfolded proteins leave the ER to undergo ubiquitin-mediated proteasomal degradation, known as ER-associated degradation (ERAD) [2]. The ER is also tightly linked to cellular calcium homeostasis [3], since ER luminal calcium binding proteins comprise the major intracellular calcium reservoir [4]. The ER is also a major cellular compartment of lipid biosynthesis including cholesterol, phospholipids, triglycerides, and sterols, for the assembly of very low density lipoprotein (VLDL) in hepatocytes [5], and for lipid droplet formation in adipocytes [6].
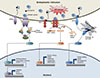
ER homeostasis cannot be maintained if physiological or pathological insults disturb protein-folding processes, resulting in ER stress [478]. A variety of stimuli can trigger ER stress, including folding-defective mutations, disturbance of metabolic and calcium homeostasis, viral infection, inflammation, hypoxia, and oxidative stress. To resolve ER stress, the ER activates an elaborate adaptive response by triggering several signal transduction cascades collectively known as the unfolded protein response (UPR) [49]. The UPR was first discovered in yeast for a single UPR signaling pathway based on the ER transmembrane protein inositol-requiring enzyme 1p (IRE1p) [1011]. However, the mammalian UPR is more diverse, consisting of at least three major signaling cascades from three ER transmembrane proteins: IRE1, which exists in α and β forms; PKR-like ER kinase (PERK); and activating transcription factor 6 (ATF6) (Fig. 1). It is generally believed that a primary short-term response to cope with ER stress should be an acute decrease in rate of protein translation to prevent further accumulation of proteins in the lumen of the ER. For the longer-term response, UPR pathways rely on transcriptional mechanisms, typically inducing the expression of target genes encoding chaperones that increase ER protein-folding fidelity, and proteins that are essential for the ERAD machinery. If the activation of these UPR pathways is not enough to alleviate ER stress, chronic UPR initiates cellular apoptosis [12].
ENDOPLASMIC RETICULUM STRESS AND UNFOLDED PROTEIN RESPONSE
Under normal circumstances, these transmembrane ER stress sensors and their signal transducers are inert due to their association with the major luminal ER chaperone, immunoglobulin heavy chain BiP (also known as glucose-regulated protein 78). During ER stress, BiP dissociates from the luminal domains of IRE1, PERK, and ATF6 to bind to unfolded or misfolded ER luminal proteins, which lead to the activation of the UPR [478]. In addition to the dissociation of BiP from these three transmembrane proteins, there are other UPR sensors and mechanisms proposed. For example, the luminal domain of IRE1β itself may directly interact with unfolded or misfolded proteins; the glycosylation of ATF6 is profoundly decreased during ER stress; or thioredoxin-interacting protein (TXNIP) regulates ER stress via protein disulfide isomerase (PDI) activation [13].
IRE1 is a single ER transmembrane protein that contains an ER luminal domain sensing unfolded or misfolded proteins, as well as a cytoplasmic domain with both intrinsic kinase activity and endoribonuclease (RNase) activity. In homeostatic conditions, kinase and RNase activities of IRE1 are inactive due to its association with BiP. Upon a variety of stress conditions, free IRE1α forms homodimers or oligomers, which increases autophosphorylation of the cytoplasmic domain. This eventually triggers its specific RNase activity, which results in an unconventional splicing process that removes 26 nucleotides from the X-box binding protein 1 (XBP1) mRNA. Converting the unspliced form of XBP1 mRNA (XBP1µ) into the shorter spliced form of XBP1 mRNA (XBP1s), leads to the efficient translation of XBP1s protein. It acts as a basic leucine zipper (bZIP) transcription factor to induce nuclear target genes encoding ER chaperones, ERAD components, lipid and phospholipid biosynthetic enzymes, and inflammatory components [8]. Moreover, with its nonspecific RNase activity, IRE1 promotes the degradation of ER-localized mRNAs, a process called a regulated IRE1-dependent decay (RIDD) of mRNA, which contributes to the reduction of protein loading in the ER. mRNA substrates cleaved by RIDD mechanism include genes involved in the lipid metabolism and specific microRNAs that derepress translation of proapoptotic genes [14]. IRE1 also activates c-jun N-terminal kinase (JNK) and IκB kinase (IKK) by recruiting the scaffold protein tumor necrosis factor receptor-associated factor 2 (TRAF2) and the apoptosis signal-regulating kinase 1 (ASK1) [1516]. JNK and IKK are two major kinases to induce Ser phosphorylation of insulin receptor substrate 1. This results in inhibition of its Tyr phosphorylation by insulin receptor, which leads to insulin resistance (IR) [1718]. There are two mammalian homologs of yeast IRE1p: IRE1α is ubiquitously expressed in cells and tissues but expression of IRE1β is limited to the epithelium of the gastrointestinal tract where it plays an important role in dextran sodium sulfate (DSS)-induced colitis probably via contributing to efficient protein folding and secretion of mucin2, a major secretory protein in goblet cells [1920].
ATF6 is an ER transmembrane protein with a cytoplasmic transcription activator domain and an ER luminal domain that senses protein-folding status. Upon ER stress, ATF6 dissociates from BiP and moves from the ER to the Golgi apparatus where it is cleaved by the mechanism of regulated intramembrane proteolysis (RIP) performed by the two serine proteases, site 1 and site 2 proteases (S1P and S2P, respectively). The cleaved cytoplasmic domain is then translocated into the nucleus, where it acts as a bZIP transcription factor to regulate expression of XBP1 and other genes involved in ER chaperones, ERAD components, protein foldases, and C/EBP homologous protein (CHOP) [48]. CHOP also acts as a transcription factor that induces the expression of genes encoding the growth arrest and DNA damage-inducible protein (GADD34), a regulatory subunit of protein phosphatase 1 (PP1), and ER oxidoreductin 1 (ERO1) [1221]. There are two Atf6 genes, Atf6α and Atf6β/creb-rp/g13 [22]. It is of interest to note that the same mechanism of RIP can cleave other ATF6 homologs with relatively less known functions. These include cAMP-response element-binding protein H (CREBH), cAMP-response element-binding protein 4 (CREB4), old astrocyte specifically induced substance (OASIS), and Box-binding factor-2 human homolog on chromosome 7 (BBF2H7) [23242526].
Under ER stress, BiP dissociation also allows PERK homodimerization and subsequent autophosphorylation, activating its cytoplasmic kinase domain. Activated PERK is able to phosphorylate the α subunit of eukaryotic initiation factor 2 (eIF2α) at Ser 51, which inhibits the activity of the guanine nucleotide exchange factor eIF2B. Thus, eIF2α phosphorylation rapidly decreases the initiation of mRNA translation; thereby, reducing the load of newly synthesized proteins in the ER. Paradoxically, the PERK-eIF2α pathway facilitates translation of ATF4, which increases transcription of genes involved in amino acid synthesis and apoptosis, such as tribbles homolog 3 and CHOP [4827]. Subsequently, CHOP is able to increase the expression of GADD34 that targets PP1 to eIF2α for its dephosphorylation. Thus, ATF4 provides a negative-feedback loop to control PERK-eIF2α signaling cascades [12]. It is now appreciated that both the ATF6 and PERK pathways trigger apoptosis by increasing CHOP expression [28]. It is worth emphasizing that, in addition to its response to ER stress, eIF2α is also phosphorylated by three other kinases: double-stranded RNA (dsRNA)-activated protein kinase (PKR), general control non-derepressible kinase 2 (GCN2), and heme-regulated inhibitor kinase (HRI), that are respectively activated by dsRNA, amino acid deprivation, and iron/heme deprivation. Because of its activation by various stresses, the eIF2α-ATF4 pathway has been designated as the “integrated stress response” [1729].
ENDOPLASMIC RETICULUM STRESS CAUSES HEPATIC INSULIN RESISTANCE
IR is developed by numerous and diverse underlying causes. Recent evidence indicates that the ER is associated with both the development of IR and its progression to type 2 diabetes mellitus [130]. One of the mechanisms by which ER stress contributes to the development of hepatic IR is the activation of transcription factors that regulate the expression of genes involved in gluconeogenesis. However, depending on which UPR pathway is activated, gluconeogenesis could be enhanced or compromised. For example, activation of CREBH, an ATF6 homolog upon ER stress primarily in the liver increases the expression of the gluconeogenic genes phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6 phosphatase (G6pase), along with inflammatory markers such as C-reactive protein [31]. By contrast, generating XBP1s by ER stress-mediated IRE1 pathway induces ubiquitin-mediated proteasomal degradation of forkhead box O1 (FoxO1), resulting in reduced gluconeogenesis [32].
Hepatic IR by ER stress may also be the result of increased lipogenesis, which leads to intracellular accumulation of diacylglycerol (DAG) and ceramide. DAG accumulation shows toxic effects for the hepatocytes and is pivotal for ER stress to cause IR and hepatic steatosis. In this regard, exposure of HepG2 cells to hyperglycemia or the saturated fatty acid (FA) palmitate, induces ER stress and activates the PERK-eIF2α pathway [33]. This in turn activates the transcription factor sterol regulatory element binding protein (SREBP)-1c to increase lipogenesis and lipid accumulation. This pathway may also contribute to ER-stress-induced hepatic steatosis through the increased expression of hepatic VLDL receptor, which promotes lipoprotein delivery to the liver [34]. The mammalian target of rapamycin complex 1, a key regulator of SREBP-1c expression, also activates ER stress, which increases hepatic IR and lipid accumulation [35]. An additional mechanism by which ER stress activates SREBP-1c processing includes rapid degradation of its upstream inhibitor insulin-induced gene 1 (Insig1) via RIDD mechanism [36].
A previously dominant hypothesis that hepatic IR is secondary to ER stress-mediated lipogenesis has been challenged by a relatively recent hypothesis that fat accumulation per se does not induce IR. The latter hypothesis is supported by the findings that the CHOP knockout (KO) mice showed normal glucose tolerance and insulin sensitivity despite marked obese phenotype [3738]. This discrepancy seems to be due to diminished inflammation in fat and liver tissues of CHOP KO mice, reinforcing the idea that IR is not induced by fat accumulation per se, but rather by inflammatory signaling cascades controlled by CHOP. By contrast, it has been shown that ER stress-mediated PERK activation may decrease insulin responsiveness through phosphorylation of FOXO1 in hepatocytes [39]. Since insulin signaling reduces FOXO1 activity via Akt and promotes insulin responsiveness, it has been suggested that inhibition of PERK might improve insulin signaling in the liver.
Fibroblast growth factor 21 (FGF21), an endocrine hormone predominantly secreted by the liver has a broad range of effects on metabolism. Recent studies indicate that either ER stress or autophagy deficiency induced FGF21 expression via the IRE1α-XBP1 or PERK-ATF4 pathway, respectively [4041]. Moreover, diet-induced obesity led to decreased autophagic activity by downregulating autophagic gene expression, and to reduction in the activity of the proteasome in the liver. This was in turn associated with activation of chronic UPR, hepatic IR, and increased glucose production. Conversely, administration of recombinant FGF21 peptide or restoration of autophagic activity in the liver of obese mice alleviated tunicamycin-induced ER stress and steatosis, and improved both hepatic insulin action and systemic glucose tolerance. As FGF21 exerts beneficial effects on lipid metabolism through counteracting ER stress, it is possible that it may display antiobesity and antidiabetic activity.
THE NUCLEAR RECEPTOR LIVER RECEPTOR HOMOLOG-1 RESOLVES ENDOPLASMIC RETICULUM STRESS AND IMPROVES INSULIN RESISTANCE IN THE LIVER
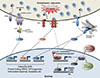
The nuclear hormone receptor liver receptor homolog-1 (LRH-1, also known as NR5A2) is expressed in tissues of endocrine origins including liver, pancreas, and intestine as well as reproductive tissue such as ovary [42]. LRH-1 plays a pivotal role in pathways that appear quite distinct from ER stress, including bile acid homeostasis, embryonic development and pluripotency of embryonic stem cells, and intestinal steroidogenesis [43]. However, a potential link between LRH-1 and ER stress resolution was raised by the observation that LRH-1 activation improves type 2 diabetes mellitus by alleviating nonalcoholic fatty liver disease (NAFLD) [44]. This was confirmed by our recent study showing that LRH-1 is necessary for resolution of acute ER stress (Fig. 2) [45]. This is dependent on a novel pathway in which ER stress-dependent LRH-1 activation induces expression of polo-like kinase 3 (Plk3) gene, which leads to increased phosphorylation of ATF2. Liver-specific Lrh-1 KO (Lrh-1LKO), Plk3 KO mice, and mice with decreased ATF2 activity were all defective in ER stress resolution upon tunicamycin challenge. Interestingly, restoring PLK3 expression in Lrh-1LKO mice was sufficient to resolve ER stress, demonstrating that PLK3 is an important LRH-1 target gene in ER stress resolution. As expected, treatment with an LRH-1 agonist protected primary hepatocytes against the toxic effects of strong UPR activation. The molecular mechanisms for how ER stress activates LRH-1 and how ATF2 contributes to ER stress resolution remain to be determined. At this stage, however, it is intriguing that the LRH-1-PLK3-ATF2 signaling pathway for ER stress resolution seems to be independent of the three canonical UPR pathways. More broadly, these studies suggest that targeting LRH-1 may provide a new therapeutic intervention in not only IR and type 2 diabetes mellitus, but also other human diseases caused by chronic ER stress.
OTHER NUCLEAR RECEPTORS CAN RESOLVE ENDOPLASMIC RETICULUM STRESS & INSULIN RESISTANCE IN THE LIVER
Several other members of the nuclear receptor superfamily affect ER stress and UPR pathways in liver, as well as in other tissues. Increased hepatic gluconeogenesis is one of the hallmarks of IR and type 2 diabetes mellitus. Particularly, glucocorticoid receptor (GR, also known as NR3C1) potently activated by endogenous glucocorticoids or a synthetic agonist dexamethasone is known to markedly increase hepatic gluconeogenesis by inducing the transcription of G6Pase and PEPCK genes [4647].
Earlier results have shown that the orphan nuclear receptor estrogen-related receptor γ (ERRγ, also known as NR3B3), a gluconeogenic transcription factor, is linked to ER stress [48]. This was supported by the observation that ERRγ can also induce expression of both ATF6 [49] and the truncated form of CREBH that is activated upon ER stress [50]. The central role of ERRγ is reinforced by the observation that it is required for induction of CREBH in response to the ER stress inducer tunicamycin [50]. It is likely that the antidiabetic effects of the ERRγ inverse agonist GSK5182 [51] are due to inhibition of these ER stress related responses as well as direct suppressive effects on gluconeogenic gene expression.
A recent report has linked increased NAFLD in the livers of aged mice to downregulation of farnesoid X receptor (FXR, also known as NR1H4) signaling induced by ER stress [52]. The chemical chaperone tauroursodeoxycholic acid, a poor agonist ligand for FXR in vitro, significantly increased hepatic FXR expression and decreased lipogenic gene expression in aged mice. ER stress inducers such as tunicamycin or thapsigargin also decreased expression of FXR and its downstream target genes, small heterodimer partner (SHP, also known as NR0B2) and bile salt export pump (BSEP, also known as ABCB11). The decrease in FXR expression was attributed to ER stress-mediated inhibition of hepatocyte nuclear factor 1α (HNF1α) transactivation of the FXR promoter [52].
Decreased activity of peroxisome proliferator-activated receptor α (PPARα, also known as NR1C1) via gene KO or perturbation of its signaling by feeding high-fructose diet impairs mitochondrial FA oxidation and increases hepatic mitochondrial stress [53]. Decreased PPARα activity was also linked to compromised expression of the sarco/endoplasmic reticulum calcium ATPase (SERCA) and to induction of ER stress and hepatic steatosis. In contrast, treatment of high-fructose diet-fed rats with the PPARα agonist WY-14643 increased FA oxidation and strongly decreased both steatosis and markers of ER stress. In another study of high-fat diet-fed mice, treatment with the selective PPARα agonist fenofibrate completely reversed glucose intolerance and hepatic IR but did not decrease either hepatic steatosis or inflammatory signaling associated with JNK or IKK [54]. This seems to be attributed to sequestration of DAG, inhibitors of insulin signaling, into the lipid droplet/ER compartment and away from the plasma membrane. Overall, it is quite likely that increased hepatic FA oxidation contributes to the documented antidiabetic effects of fibrates in human clinical trials [5556], and this may contribute to decreased ER stress.
ER stress-activated indicator transgenic mice in which green fluorescent protein (GFP) expression is induced under ER stress conditions were used to examine whether pioglitazone, a selective PPARγ (also known as NR1C3) agonist, improves ER stress in vivo [57]. In high-fat and high-sucrose diet-fed transgenic mice, GFP was clearly decreased as early as 4 weeks after initiation of pioglitazone treatment, prior to the improvement of IR. This study suggests that the antidiabetic effects of pioglitazone may be at least due to reducing ER stress in the liver. Other PPAR agonists also link their antidiabetic effects to decreased ER stress. Thus, novel dual agonists for PPARα/γ showed antidiabetic effects associated with improving fatty liver, inflammation and ER stress in metabolic tissues of leptin receptor (Lepr)-deficient db/db mice [5859].
Finally, liver X receptors (LXRα and β, also known as NR1H3 and NR1H2, respectively) agonist treatment has been reported to decrease saturated FA-induced ER stress in primary hepatocytes by inducing expression of the remodeling enzyme lysophosphatidylcholine acyltransferase 3 (Lpcat3), which modulates membrane phospholipid composition by increasing incorporation of unsaturated FA chains [60]. Although direct studies of LXR activation in vivo are complicated by its potent lipogenic effect, Lpcat3 overexpression has been shown to be sufficient to ameliorate ER stress and also improve glucose homeostasis in livers of both ob/ob and db/db mice.
CONCLUSIONS
In this review, we have briefly described current understandings of how ER stress activates UPR pathways, and of how chronic ER stress causes peripheral IR. In addition, we have described how nuclear receptors contribute to ER stress resolution and ER stress-mediated apoptosis in different metabolic tissues with an emphasis on the aspects of nuclear receptor LRH-1 in hepatic ER stress resolution, which seems to be independent of canonical three branches of UPR pathways. It would be of great interest to further investigate why the intact activation of each canonical UPR signaling could not be sufficient for resolving tunicamycin-mediated ER stress in Lrh-1LKO mice. It would also be of interest to understand how LRH-1/PLK3/ATF2 pathway could integrate into canonical UPR signaling pathways. Lastly, it would be necessary to further understand the roles of nuclear receptor LRH-1 upon other kinds of physiologic or pathologic ER stresses, which might provide a therapeutic strategy to overcome diverse metabolic diseases.
 XML Download
XML Download