

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Of the numerous complications associated with diabetes, cardiovascular diseases (CVD) remain the major cause of death [1]. In both type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM) there is a complex milieu of systemic changes including hyperlipidemia and hyperglycemia that contribute to CVD risk [234]. This increased prevalence of heart failure in the absence of coronary artery disease and hypertension is often referred to as diabetic cardiomyopathy [5]. Typically, the healthy heart shows a remarkable capacity to utilize lactate, ketones, fatty acids, and glucose in a concentration-dependent manner [6]. This flexibility in substrate utilization is developmentally significant, as it is seen at birth when the mammalian fetal heart switches from a reliance on lactate and glucose to one of fatty acid utilization [7]. It has long been known that in the case of obesity and diabetes, progression to heart failure is often seen as a result of excess nutrient supply, insufficient nutrient utilization, dysfunctional nutrient storage and oxidation, or a combination of the above [8]. The detriment of excess nutrient availability towards lipotoxicity, glucotoxicity, and glucolipotoxicity has all been explored as contributing factors to cellular dysfunction in diabetes [910]. Evidence continues to point to a central role for metabolic dysfunction in disease progression and continued progress has been made at defining the mechanisms of action. Candidate mechanisms of diabetes-induced dysfunction include: (1) increased reactive oxygen species (ROS); (2) increased advanced glycation end products (AGEs); (3) increased polyol flux; (4) increased protein kinase C (PKC) activation; (5) increased protein O-linked N-acetylglucosamine (O-GlcNAc); and (6) altered gene expression [1112]. Progress on deciphering each of these metabolic perturbations in the development of diabetic complications has been made and recently reviewed in detail [13]; the current review will highlight some of these mechanisms in relation to glucose.
CARDIAC GLUCOSE UTILIZATION IN DIABETES
How glucose metabolism is altered in diabetes
The mammalian fetal heart relies primarily on lactate and glucose utilization, a metabolic phenotype that is quickly reprogrammed at birth with the introduction of milk into the diet and throughout development to an adult heart that relies predominantly on fatty acid oxidation [7]. Glucose utilization serves as the major carbohydrate that accounts for 10% to 20% of myocardial high energy phosphate production in the healthy heart. For the most part the heart can utilize metabolic substrates in a concentration and delivery specific manner. However, for more than 60 years, researchers have known that despite excess circulating glucose levels, the diabetic heart shows a preferential oxidation of fatty acids which is in stark contrast to the hypertensive heart that reverts to glucose utilization [8]. The increased reliance on fatty acid oxidation results in higher costs in mitochondrial oxygen consumption in the diabetic heart and is believed to contribute to ventricular dysfunction. Impaired glucose utilization in diabetic myocardium is mediated in part by reduced glucose uptake, reduced glycolytic activity, and reduced pyruvate oxidation. Reduced glucose transport across diabetic myocardium has been ascribed to decreased expression and function of members of the solute carrier family 2A which encode the glucose transporters (GLUTs), of which seven have been reported to be expressed in the heart (GLUT1, 3, 4, 8, 10, 11, and 12) [14]. The two best characterized in heart failure and diabetes, specifically, are GLUT1 and 4 [14]. Although the other five members have relatively lower expression, understanding their contributions to cardiac hexose utilization may help unravel some of the mystery pertaining to decreased glucose uptake but increased flux through downstream pathways in the context of diabetes. In uncontrolled T1DM, reduced glucose uptake is due to insufficient insulin supplement; in the more prevalent T2DM, impaired stimulation of glucose transport in response to insulin is due to insulin resistance of the myocardium [15]. Furthermore, the interaction between glucose uptake and other metabolic pathways could result from disproportionate changes in glycolysis and glucose oxidation, an area that needs to be further defined. For example, elevated fatty acids have been proposed to induce myocardial insulin resistance and reduce GLUT4 expression in diabetic hearts [16]. However, a number of studies have also reported that cardiac tissue from T2DM patients [17] and in rodent models [18] respond normally to insulin, and show regular glucose uptake, suggesting that despite insulin resistance, T2DM hearts can still be responsive to insulin. Although considerable work has focused on the role of fatty acids as a causal factor for myocardial insulin resistance in diabetes, the molecular mechanisms involved are incompletely understood. Moreover, the disrupted metabolic substrate milieu and other comorbidities associated with animal models of diabetes such as hyperglycemia, hyperinsulinemia, hyperlipidemia, and elevated hormone levels make it difficult to decipher the mechanisms of fatty acid-induced insulin resistance in the heart.
Following its transport into cardiomyocytes, glucose is broken down through glycolysis to yield pyruvate that has three known metabolic fates: carboxylation to oxaloacetate or malate, reduction to lactate, and decarboxylation to acetyl-CoA. In the third step, pyruvate is decarboxylated to form acetyl-CoA through pyruvate dehydrogenase (PDH), a multienzyme complex located in mitochondrial matrix. In diabetes, cardiac glycolysis and pyruvate oxidation both are reduced as evidenced from animal models [1920]. Increased fatty acid oxidation leads to elevation of citrate concentration that reduces the activity of phosphofructokinase, an enzyme that converts fructose 6-phosphate to fructose 1,6-bis-phosphate. In addition, increased cardiac fatty acid oxidation is thought to inhibit PDH activity in diabetes by inducing pyruvate dehydrogenase kinase (PDK) that phosphorylates and deactivates PDH complex. A recent study in a rat model of T2DM has demonstrated that administration of PDK inhibitor restored the myocardial PDH activity and subsequent improvement in diastolic function [21]. Also, a mouse model with cardiac deficiency of malonyl-CoA decarboxylase, an enzyme promoting mitochondrial fatty acid oxidation, demonstrates that reduced fatty acid oxidation restored cardiac glucose oxidation in mice on high fat diet [22]. These data further suggest a strong influence of fatty acid oxidation over cardiac glucose utilization in diabetic hearts. Overall, myocardial glucose utilization is reduced during diabetes that appears to take place at multiple levels, including reduced glucose transport, suppressed glycolysis and reduced PDH activity by the PDKs.
How cardiac function is altered by glucose in diabetes
In the previous section we discussed how overall glucose uptake and utilization is compromised in diabetic heart, this section will focus on how glucose itself impacts the heart. Independent of systemic hyperglycemia, reduced glucose utilization is a metabolic signature of diabetic hearts, and it is believed to be one of the major causes of loss in metabolic flexibility leading to pathogenesis of diabetic cardiomyopathy. Despite reduced glucose oxidation, several studies provide experimental evidence that hyperglycemia can independently mediate the detrimental effects of diabetes through activation of alternative pathways of glucose metabolism including; the polyol pathway, increased AGEs, oxidative stress, mitochondrial dysfunction, and increased activity of PKC. In this section, we will discuss the role of glucose in the progression of diabetic cardiomyopathy through these pathways. It is apparent that with reduced myocardial glycolysis and glucose oxidation in diabetes, there is a mismatch between glucose availability in the circulation and its utilization by the heart, which still exposes the heart to significant levels of glucose. It is essentially a state of mismatch between demand versus supply, where the heart has an excess supply of nutrients than it needs for adenosine triphosphate (ATP) generation. Reduced glycolytic activity in the diabetic heart leads to accumulation of intermediates of glucose catabolism, which can participate in several non-ATP producing pathways essential for regulating cell growth and function. In the healthy heart, when intracellular glucose is converted to glucose-6-phosphate (G-6-P), the majority of it enters the glycolytic pathway and is metabolized to pyruvate, which is further oxidized in the mitochondria. In addition to glycolysis, G-6-P is also a substrate for glycogen synthesis, the pentose phosphate pathway, and the aldose reductase/polyol pathway that may have implications in diabetic complications. Increased glucose flux through these pathways has been reported to be associated with oxidative stress and the development of microvascular and cardiovascular complications [23]. In addition to above metabolic pathways, fructose-6-phosphate generated from glycolysis can enter the hexosamine biosynthetic pathway (HBP), increased uridine diphosphate-GlcNAc, and posttranslational modification of nuclear, cytoplasmic, and mitochondrial proteins via O-linked attachment of β-N-acetyl-glucosamine to serine or threonine residues. The latter is regulated with addition by O-GlcNAc transferase and removal by O-GlcNAcase. A growing body of evidence strongly suggests the causative role of aberrant cardiac O-GlcNAc signaling in mediating the adverse effects of diabetes on the heart [24]. Moreover, elevated glucose levels due to chronic hyperglycemia can non-enzymatically glycate proteins to form AGEs, which not only are directly damaging but they also contribute to ROS generation. AGEs are formed when glucose or glucose metabolites produce stable, covalent modification of proteins. AGEs have been reported to be involved in the pathogenesis of diabetic cardiomyopathy [25]. Taken together, experimental evidence from rodents and human, it is apparent that even overall glucose utilization is compromised in diabetic myocardium, high glucose associated with chronic hyperglycemia can independently contribute to myocardial dysfunction in diabetes beyond its role as an efficient metabolite for the generation of ATP. Although all these alternative metabolic pathways individually contribute to diabetic complications, the exact path linking high glucose-induced damage to the heart remains elusive. However, compared to other hyperglycemia-induced non-ATP producing glucose catabolic pathways, the induction of HBP is now well established as an important contributing to the progression of cardiac dysfunction in diabetes.
Glucose and mitochondrial function
As both fatty acids and glucose contribute to the mitochondrial oxidative phosphorylation for ATP generation, it is apparent that altered energy metabolism associated with diabetes can directly impact mitochondria in diabetic myocardium. Diabetes-induced mitochondrial dysfunction has been described as a cause of cardiac dysfunction as evidenced animal models diabetes mellitus [26]. While cardiac dysfunction has been well documented in both T1DM and T2DM rodent models, mitochondrial dysfunction may be affected differently in both type of diabetes. A recent report by Marciniak et al. [27], has shown that cardiac mitochondrial function was unchanged in the streptozotocin-induced model of T1DM suggesting differential association of mitochondrial dysfunction with cardiac dysfunction in both types of diabetes. Oxidative stress is considered one of the major factors that govern onset and progression of diabetes. Mitochondria are a major source of ROS production, and increased ROS production during diabetes results from excess fatty acid-mediated availability of electrons delivered to the electron transport chain. Hyperglycemia is also believed to be a major contributor of mitochondrial dysfunction in diabetic heart. Seminal work by Brownlee [28] has focused on the role of hyperglycemia mediating their detrimental effect on CVD associated with diabetes mellitus. Major molecular mechanisms that have been proposed to mediate high glucose-induced mitochondrial damage include: increased glucose flux through polyol pathway and formation of AGEs, activation of PKC, and increased hexosamine pathway. However, activation of polyol pathway and induction of PKC have been mainly described as mechanisms inducing damage in microvasculature. Over the past few years, HBP flux has emerged as a major player responsible for mitochondrial dysfunction in diabetic myocardium. By using neonatal rat ventricular cardiomyocytes treated with high glucose, Hu et al. [29] have first showed that increased mitochondrial protein O-GlcNAcylation blunts mitochondrial function. More recently, two recent reports have determined the complete cardiac mitochondrial GlcNAc proteome and provide evidence that disruption of O-GlcNAc cycling is associated with cardiac mitochondrial dysfunction in diabetes [2430]. These results highlight the importance of altered myocardial O-GlcNAcylation regulating mitochondrial function in diabetic heart. While these results from rodent models establish the importance of hexosamine flux and alternative glucose pathways in diabetic cardiomyopathy, clinical significance of these pathways, and the specificity of their regulation, remain to be determined.
Glucose and gene expression
In the prior sections we have focused on direct regulation of proteins for metabolism and signaling. It has also been proposed that diabetes reprograms the cardiomyocyte to further exacerbate disease progression. Growing evidence supports that part of this reprogramming is directly mediated by glucose-induced gene expression changes; either by direct GlcNAcylation of transcription factors and the histone core or indirectly by influencing other gene expression and epigenetic machinery [31]. The clinical significance of changes in glucose utilization relative to long-lasting effects is supported by studies of both T1DM and T2DM patients [3233]. The increased risk of CVD and heart failure in these patients in response to either high or low glucose levels persists for years, suggesting that the mechanism involves more than just delivery of substrate to mitochondria. One explanation for the growing prevalence of these diseases and the associated CVD is the role of epigenetics in regulating gene expression. It is now clear that changes in environment, diet, and activity levels all modify gene expression in an organism and that of its offspring without altering the genomic sequence. The contribution of epigenetics to CVD was first suggested by findings of the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complication study [34]. Specifically, patients who were continuously on intensive therapy with tight glycemic control, had a significantly lower incidence of cardiovascular complications than patients who were initially on conventional therapy and subsequently switched to intensive therapy [353637]. The benefit of an initially lower glycosylated hemoglobin level resulted in reduced progression of CVD that persists over 20 years later [33]. Similarly a long-term follow-up study of T2DM patients assigned tight glycemic control revealed fewer major cardiovascular events, though mortality was the same [32]. Supporting that although survival was similar between groups, there is a clear link between glucose levels and cardiovascular outcomes [38]. These findings support a unified hypothesis that prior changes in glucose levels may lead to persistent molecular changes predisposing patients to the development of cardiovascular complications in a process termed "glycemic memory." Epigenetics is a rapidly expanding area of study that includes expression of non-coding RNA (e.g., microRNA and long-non-coding RNA), posttranslational modifications to histones (e.g., acetylation and O-GlcNAcylation), and DNA (e.g., 5-cytosine methylation and 5-hyrdoxymethylcytosine). The contribution of glucose to the regulation of multiple cell types in the cardiovascular system was reviewed elsewhere earlier this year [39] in addition to the established role of regulating transcription factors [40]. However, more work needs to be done on if these same mechanisms are directly impacting the cardiomyocyte.
Glucose and contraction
Heart failure that accompanies diabetes is multifactorial, including contractile dysfunction due to reduced cardiomyocyte Ca2+ transients. This change in sensitivity change has recently been shown to be glucose-mediated through increased O-GlcNAcylation of cardiac contractile proteins resulting in decreased Ca2+ sensitivity, and may contribute to the contractile dysfunction noted in diabetic rats [41]. Interestingly this change appears to be from both increased O-GlcNAc as well as a distinct subcellular redistribution of this glycosylation. Further evidence of a direct role of glucose on regulating contraction comes from demonstrated reductions in sarco/endoplasmic reticulum Ca2+ ATPase (SERCA2, encoded by ATP2A2), a crucial mediator of cardiac function in various etiologies of cardiac dysfunction, since its activity is required for the re-sequestering of Ca2+ during diastolic relaxation [4243]. Similarly, it has been shown via RNA-sequencing analysis that gene expression of contractile machinery is significantly disrupted in heart failure, further substantiating the notion that SERCA2 is involved in the cardiac dysfunction found in diabetic patients [4244]. Clinical trials are underway to determine whether adeno-associated virus isotype 1 mediated restoration of SERCA2 expression in human heart failure increases functional cardiac capacity and alleviates symptoms of hemodynamic insufficiency [45]. It is expected that increasing SERCA2 activity in the heart will restore contractility, as similar animal interventions have attenuated hepatic pathologic markers in diabetes [46].
In addition to the direct role of SERCA2 regulation, stromal interaction molecule 1 (STIM1) induction has also been proposed as a central player in the pathogenesis of diabetic heart disease due to its role in Ca2+ sensitivity [47]. Previous studies have shown that STIM1 can be posttranslationally modified by O-GlcNAcylation to attenuate Ca2+ entry in diabetic hearts [48], although the clinical significance of these observations is currently unknown.
TARGETING GLYCEMIC CONTROL IN HEART DISEASE
Currently, the pharmacologic approach to managing diabetic heart failure patients is a concerted attempt to treat both the diabetic complications and the cardiac dysfunction as separate diseases. However, while the treatment of cardiac dysfunction in this patient population is similar to that of non-diabetic heart failure, particular consideration must be taken in addressing other end-organ effects of chronic hyperglycemia, particularly diabetic nephropathy. For this reason, appropriate medications include those with pharmacodynamic properties independent of renal function.
Numerous clinical trials have considered the possibility of cross-talk between the management strategies of diabetes mellitus and heart failure, particularly in the context of insulin-sensitizing medications. Although the use of thiazolidenediones to manage blood glucose levels correlates with a reduced relative risk of heart failure [49], its use in diabetic patients has been associated with increased mortality if initiated in patients who have already been diagnosed with heart failure [50]. In contrast, the use of metformin has traditionally been considered an effective management diabetic patients with heart failure [51]. However, its use is contraindicated in the setting of acute decompensated heart failure, when renal perfusion falls below adequate levels for renal excretion of metformin, increasing risk of mortality due to metformin-associated lactic acidosis [52].
Beyond the use of metformin as an effective insulin-sensitizing monotherapy, multiple clinical trials have been performed to evaluate the glycemic efficacy of dipeptidyl peptidase-4 (DPP-4) inhibitors as adjunctive therapy, which showed great promise as anti-diabetic treatments for heart failure patients. DPP-4 inhibitors (colloquially termed the “-gliptins”) block the DPP-4 mediated enzymatic degradation of glucagon-like peptide-1, which in turn promotes glucose-dependent insulin release from the beta cells of pancreatic islets. However, it has been shown that the use of these drugs did not improve mortality in heart failure patients when used in combination with a polypharmacy approach of current best treatments [53]. Furthermore, the use of DPP-4 was associated with an increased risk of hospitalization for heart failure in subjects with T2DM who had or T2DM patients with existing CVD [5354].
ADA recommended not to use 'diabetic' as a meaning of adjective for patient with diabetes. An alternate method of lowering blood glucose levels lies in reducing the reabsorption of glucose from glomerular ultrafiltrate in the kidneys. The reabsorption of glucose, coupled to that of sodium, is performed by the appropriately-named sodium-glucose transporter isoform-2 (SGLT2). The inhibition of this transporter is a noteworthy class of anti-hyperglycemic medications, termed the “gliflozin” drugs, that effectively lower blood glucose by reducing the reabsorption of glucose in the renal tubules, particularly when given with metformin [55]. Despite increasing the risk of urinary tract infections, the use of SGLT2 inhibitors has been shown to improve outcomes in patients with T2DM who had or T2DM patients with simultaneous CVD [56].
CONCLUSIONS
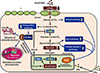
The healthy heart maintains a balance in substrate utilization, primarily in a concentration dependent manner. In diabetes this flexibility is lost and the heart is no longer able to readily switch between mitochondrial substrates. The resulting build-up of these metabolic intermediates spills over into other pathways resulting in aberrant protein posttranslational modifications, disrupting signaling, a reprogramming of gene expression, and altered contractile machinery and sensitivity to calcium (Fig. 1). The end result is that subjects with diabetes have a 67% chance of dying from a CVD [1]. The current review only focused on the direct impact of glucose on the heart. It is important to note that diabetes is a disease of multiple metabolites and many others have recently reviewed the changes in fatty acids and ketone bodies in CVD [5758]. Furthermore, diabetes has profound effects on the vasculature to impact CVD and outcome, topics that have also been recently reviewed in detail [59]. In closing, it will take a complete understanding of the above combined with the sweet side of diabetes to understand and hopefully reverse the dysfunction seen in the broken diabetic heart.
 XML Download
XML Download