

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
All international and national health organizations report that obesity has doubled since 1982. In 2014 over 1.9 billion adults were overweight and of these over 600 million were obese worldwide. This makes obesity a serious pandemic and public health problem [123]. Importantly, childhood obesity has tripled in the last 30 years and the World Health Organization reports that 42 million children under the age of five were overweight or obese in 2013 [45]. The clinical relevance of obesity derives from the fact that obesity is associated with several metabolic risk factors that define the metabolic syndrome (MetS) [6]. MetS is strongly associated with the development of comorbidities, including cardiovascular diseases, diabetes as well as non-alcoholic fatty liver disease (NAFLD) [78910].
NAFLD is one of the most common causes of chronic liver disease (CLD) and liver-related morbidity and mortality worldwide. Currently, NAFLD has an estimated prevalence in the general population of 20% to 30% in Western Countries and 5% to 18% in Asia, and it is predicted to increase over time [11121314]. An additional dramatic increase of NAFLD has been associated with obesity and excessive fructose consumption [1516]. Notably, the overall prevalence of childhood NAFLD has reached about 10%, with a worrisome prevalence rate of 17% in teenagers [17]. This prevalence dramatically increases up to 40% to 70% among obese children [1819]. A subset of 3% to 5% of NAFLD patients can develop early non-alcoholic steatohepatitis (NASH) [2021], characterized by lobular and portal inflammatory infiltrates of monocyte-derived macrophages, neutrophils and lymphocytes; various degree of fibrosis, hepatocyte cell death and pathological angiogenesis. With the progression of the disease to advanced NASH, extensive fibrosis is associated with high risk of developing cirrhosis, liver failure, portal hypertension and/or hepatocellular carcinoma (HCC), the third leading cause of cancer death worldwide [2223]. Advanced fibrosis and cirrhosis are the principal causes of liver-related morbidity and mortality. In a recent study published by Wong and colleagues [24] on the annual trends of new liver transplant waitlist registrations they reported that from 2004 to 2013 NASH, as cause of CLD, showed an increase of almost 170% in the number of new waitlist registrations, compared to other CLDs such as alcoholic liver disease or hepatitis C virus. This makes NASH the second leading etiology of CLD in new waitlist registrations for liver transplant and projections show that NASH will become the leading cause of liver transplantation by 2020 [2425].
The most accepted concept on the pathogenesis of NAFLD involves multiple concomitant events or 'hits' associated with a crosstalk between environment, host genetics and gut microbiota [2627]. In this scenario, stressed or damaged cells release various dangerous signals that influence and drive NASH-related outcomes such as isolated steatosis, sterile inflammation, fibrosis, pathological angiogenesis, and progressive liver injury [2628]. In this review, we will focus and uncover novel insights on cell-derived extracellular vesicles (EVs) as damage-associated molecular signals involved in the molecular pathogenesis of NASH. We will describe studies reporting the EV-driven signaling and molecular pathways related to the initial and advanced events involved during the progression of NASH. We will describe the EV genesis from cells exposed to toxic agents and the reported effects on functionality and activity of the target cells.
EXTRACELLULAR VESICLES AND NAFLD PROGRESSION
Different populations of EVs at play
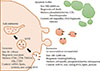
EVs (or exovesicles, as most recently named) are a heterogeneous population of small membrane-surrounded structures released by cells in the extracellular environment as well as in the bloodstream and are involved in normal physiology as well as in the pathobiology of disease [29303132]. EVs are effective communicators that are generated by a cell of origin or parenteral cell and can act on a number of target cells in an autocrine or paracrine pathway in the extracellular environment where they are released as well as in an endocrine manner, acting as long range signals [33343536]. EVs shuttle a variety of bioactive molecules that may reflect the cell of origin as well as the specific stress that induces their formation and release. They transport and transfer in target cells bioactive molecules, including mRNA, different non-coding RNAs (microRNA, longRNA mitochondrial associated tRNA, small nucleolar RNA, small nuclear RNA, Ro associated Y-RNA, vault RNA, and Y-RNA), proteins, DNA and lipids that influence and modulate various cell responses [3738]. EVs have been divided in three main categories: exosomes, ectosomes (or microparticles [MPs] or microvesicles) and apoptotic bodies (Fig. 1) [293940]. However, several reports have shown that these subpopulations have one or more characteristics in common and a clear distinction among the different categories is still not completely possible.
Exosomes are approximately 30 to 100 nm in size and originate from inward budding of the membrane of late endosoma compartments in the cytosol of cells. The fusion of endosomal compartment with the plasma membrane facilitate the release of exosomes that contain proteins, non-coding RNA (miRNA, long-RNA) and RNA interfering. Several studies have identified some exosomal membrane proteins, such as tetraspanins, CD63, CD81 and CD9 or TSG101 and Alix [3941].
Ectosomes are approximately 100 to 1,000 nm in size and are release from outward budding and blebbing of the plasma membrane directly in the extracellular space [29]. Most ectosomes express phosphatidylserine on the outer leaflet of the membrane, allowing their detection. However, a subpopulation of ectosomes does not express phosphatidylserine. For this reason different labeling approaches and cell-specific markers are used to detect this subpopulation of EVs. Ectosomes carry a variety of different bioactive molecules, such as lipid, proteins, non-coding RNA and mRNA that can be shuttled into the target cells where ectosomes are efficiently internalized and induce a variety of different cell responses [34424344].
Apoptotic bodies are the largest EVs with a diameter between 500 to 4,000 nm [45]. Similarly to ectosomes they are released by blebbing of the plasma membrane of the apoptotic cells and they express phosphatidylserine on their surface [46]. Differently from exosomes and ectosomes, apoptotic bodies carry cell organelles, DNA fragments and histones [4748]. Common markers of apoptotic bodies include complement component C3b and thrombospondin [49].
Lipotoxicity as a trigger for release of EVs
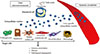
Lipotoxicity refers to a process by which accumulation of certain toxic lipids such as saturated free fatty acids (SFA), free cholesterol, or ceramide and other sphingolipid among others in hepatocytes triggers various molecular pathways of cell stress and eventually results in cell death have evolved as a key event during NAFLD progression [505152]. Elegant studies have shown that hepatocytes exposed to the SFA palmitic acid—lipotoxic lipid abundantly present in circulation of patients with NASH [5354]—produce and release large quantities of EVs that may act on various target cells and contribute to key processes involved in NAFLD pathogenesis, including immune reactions, angiogenesis, and fibrosis (Fig. 2).
EVs in liver pathological angiogenesis
Liver pathological angiogenesis consists in the development of an abnormal angioarchitecture during the progression of progressive CLDs, including metabolic-related liver diseases as NASH. The clinical relevance of pathological angiogenesis in NASH is associated with the evidence that pathological neovessel formation drives hepatic fibrogenic progression and leads to the development of cirrhosis and related complications, such as HCC [5556]. Pathological angiogenesis occurring during a CLD is primarily regulated by concomitant liver events that involved complex cell-to-cell interaction and release of various pro-angiogenic factors in the liver microenvironment [57]. Several elegant studies have reported a role of EVs in activation of endothelial cells (ECs) and promotion of angiogenesis. In our previous studies we have demonstrated that hepatocyte-derived EVs released during lipotoxicity have potent pro-angiogenic effects and promote EC activation in a Vanin-1-mediated internalization [34]. In a pioneered study by Witek et al. [58], they showed that platelet-derived growth factor BB stimulated myofibroblast-like hepatic stellate cells (HSCs) and reactive cholangiocytes, release MPs that contain active Sonic hedgehog (Shh) and Indian hedgehog (Ihh)—two of the proteins of the signaling pathway family of hedgehog. The same authors showed that plasma and bile level of Hh-containing MPs were elevated in chronic biliary fibrosis and bile duct ligation rats, respectively. MPs carrying Hh molecules induced a pro-angiogenic switch in sinusoidal ECs, followed by in vivo capillarization. In a more recent report by Lemoinne and colleagues [35], it has been demonstrated that portal myofibroblasts (PMFs) promote the formation of neo vessels triggering scarring development into the parenchyma in both CCl4-induced liver fibrosis and NASH models. Through various experimental approaches, authors showed that activated PMFs release MPs that carry and transfer the pro-angiogenic factor vascular endothelial growth factor A (VEGF-A) to ECs, inducing EC activation and tubulogenesis via VEGF-A receptor. However, the mechanism driving the release of VEGF-A by MPs was not further investigated by the authors of this published study. In conclusion, EVs play a crucial role in liver angiogenesis and may link pathological angiogenesis and fibrogenesis during the progression of NASH by transferring various agents, including growth factors, hedgehog molecules, proteins, and miRNAs.
EVs in inflammatory responses
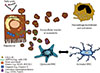
In a recent study published by Ibrahim et al. [36], they provide novel insights in the lipotoxicity-mediated release of hepatocyte-derived EVs during NASH development. Authors report that mixed lineage kinase 3 (MLK3) mediates the release of fat-laden hepatocyte-derived EVs that carry chemokine (C-X-C motif) ligand 10 (CXCL10), a potent hepatocyte-derived macrophage chemoattractant [59]. These findings were confirmed by pharmacologically inhibiting MLK3, which resulted in a significant decrease in the amount of CXCL10 released by lipid-stressed hepatocytes. These observations highlight a novel role of CXCL10-enriched EVs in monocyte-derived macrophage recruitment and activation of resident Kupffer cells (the resident hepatic macrophages) during the progression of NASH (Fig. 3). Adipose tissue has also emerged as crucial player in the development of hepatic steatosis and chronic low-grade inflammation, a key link between obesity and the associated metabolic dysregulation [33606162]. In line with this concept, one of our recent studies first-authored by Eguchi et al. [33] shows that mature differentiated adipocytes exposed to palmitic acid resulted in the release of a marked amount of EVs that are potent chemoattractant factors that induced migration of macrophages. Through various lines of evidence, we showed that the release of EVs as chemotactic signals by adipocytes treated with palmitic acid was mediated by caspase-3 activation [3334]. These findings identified adipocyte-derived EVs as novel "find-me" signals linking adipocytes stress to macrophages recruitment. The EV-mediated cell-to-cell communication is a central event in a multifactorial disease as NASH.
EVs in liver fibrogenesis
Several studies have shown that advanced fibrosis and cirrhosis are crucial pathophysiological events of CLDs and lead to clinical life-threatening consequences [63646566]. Notably, advanced fibrosis has been reported to increase the risk of HCC 25-fold [2367]. Activated HSCs are the main cell type that induce and sustain the extracellular matrix remodeling and collagen deposition during liver fibrogenesis [68697071]. Among many factors, circulating miRNAs are pivotal regulators of HSC phenotype and several studies have identified miRNAs up-regulated in experimental models of NAFLD or activated HSC in vitro [72737475]. For an extensive and complete overview on the role of miRNAs in the pathogenesis of CLDs we recommend other reviews [7677787980]. In this paragraph we will highlight some studies describing the role of EVs in liver fibrogenesis in NASH. A first study showing that HSCs can be activated by EVs was published by Kornek and colleagues [81]. Authors described the role of T cells in the fibrolytic activation of HSCs [81]. In this study it was demonstrated that activated and apoptotic CD4+ and CD8+ T cells release MPs in the extracellular environment and in circulation that fuse with HSCs and transfer membrane molecules such as CD147, triggering the upregulation of fibrolytic molecules. The biological significance of "horizontal transfer" of bioactive molecules by EVs has been demonstrated by various studies. Pro-fibrogenic connective tissue growth factor (CCN2) has been identified exosomes packaged by activated HSCs and shuttled to other quiescent or activated HSCs as form of paracrine pro-fibrogenic activation during a liver injury [82]. On the other hand, a second study showed that exosomes released by quiescent HSCs have an anti-fibrogenic potential by transferring Twist1, a transcription factor that activates miR-214 and results in CCN2 suppression [83]. The evidence that EVs can shuttle and deliver small or long non-coding RNA has sparked a great interest in the basic pathological mechanisms of liver fibrosis development. Notably, it has been demonstrated that EVs shred by hepatocytes exposed to palmitic acid shuttle miR-128-3p into HSCs and induce a down-regulation of PPAR-gamma, a central modulator of HSC quiescence (Fig. 3) [84858687]. In a complex microenvironment such as the one present during NASH-related fibrogenesis, EVs released by several cell sources play a key role in the homeostasis and balance between pro-fibrogenic and anti-fibrogenic mechanisms.
EVs as novel biomarkers to monitor liver injury in NASH
Currently, liver biopsy remains the gold standard procedure to distinguish simple steatosis from steatohepatitis. However, it is costly, invasive and carries some morbidity and a rare mortality risk [63]. The biogenesis and composition of EVs have suggested that they might be identified as novel non-invasive and reliable biomarkers for liver injury during NASH development. This concept is further supported by the evidence that EVs can be detected and isolated from a variety of biofluids, including serum and plasma samples both from experimental animal models and human patients with NASH [888990]. Level of circulating MP derived from CD14+ and invariant natural killer T (iNKT) cells were significantly elevated in patients with NAFLD/NASH, independently of alanine aminotransferase levels or histopathological markers of disease activity [91]. In line with these findings, a complete characterization of bioavailability, composition, and origin of circulating EVs would indicate whether EVs can be used to discriminate different CLDs or different disease stages during the progression of CLDs such as NASH. Experimental evidence supporting this concept has been shown in one of our recent reports. In an experimental model of diet-induced NAFLD, early- and advanced-NASH, we observed that level of blood EVs increased over time during the NASH progression and was strongly associated with some key outcomes of NASH, including the extent of liver fibrosis, angiogenesis, and cell death [92]. The bioavailability of EVs in biofluids is only one of the fascinating aspects of these effective "messengers." Indeed, the composition of EVs suggests an extraordinary potentiality for biomarker development. As a proof of this, miRNAs encapsulated in circulating EVs have increasingly exhibited great potential to fulfill several criteria for being considered a good biomarker, such as specificity, sensitivity, robustness, predictivity, translatability, and non-invasiveness [909394]. Circulating EVs isolated from murine models of experimental NASH were enriched in miR-122 and miR-192, two microRNAs abundantly expressed in the liver [9596]. The level of these microRNAs increased in EVs and decreased in livers over time during NAFLD progression [92]. The release of these microRNAs from stressed or damaged hepatocytes in EVs during NAFLD progression may provide an attractive explanation for the decreased expression level of miR-122 found in the livers of patients with advanced NAFLD [97], as well as early stage hepatocarcinogenesis from NASH in both an animal model and human tissue samples, as recently reported [9899]. In addition to microRNAs, a complete proteomics analysis of circulating EVs isolated from rodents with NASH vs. controls revealed a unique disease-specific protein signature that can be employed as "barcode" for diagnostic novel strategies in NASH [92]. These findings provide important support to the development of EV-based lab-on-a-chip devices [100] that can be used for "liquid biopsy" to introduce novel and non-invasive diagnostic strategies for NASH.
CONCLUSIONS
In conclusion, we have highlighted several of the most recent and original studies reporting the role of EVs as link between lipotoxicity, inflammation, angiogenesis, and fibrosis, all key events in the pathogenesis of NASH. Evidence shows that EVs may act as or transfer damage-associated molecular patterns to target cells of the liver microenvironment and orchestrate many of the main pathological events occurring concurrently during the development of NASH. A second important aspect of EVs consists on the fate of EVs, either benign or malignant and this seems to depend on the phenotype of the cell of origin.
Future studies will need to further (1) dissect the mechanisms of release of EVs by the parenteral cells in pathological conditions; (2) investigate additional functions of EVs during the pathogenesis of NASH; and (3) elucidate whether an impaired balance between "bad" and "good" EVs might be a potential cause of many CLDs.
 XML Download
XML Download