

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cardiovascular disease (CVD) including stroke and coronary artery disease (CAD) is the global leading cause of morbidity and mortality [1]. Diabetes mellitus (DM) is associated with accelerated atherothrombosis; consequently, DM patients have shown a 2- to 4-fold greater risk of CAD and cerebrovascular disease than non-DM patients [2]. Of note, diabetic subjects without a history of CAD have shown a similar risk of future CAD events similar to nondiabetic subjects with a history of myocardial infarction (MI) [3]. Following the first manifestation of CVD, DM patients also have a higher risk of recurrent cardiovascular complications than non-DM patients despite standard medical treatment.
Because the global prevalence of DM is increasing rapidly (e.g., 165% between 2000 and 2050), there is an unmet need to reduce the incremental burden of atherothrombotic events in these DM patients [4]. Heightened cardiovascular risk in diabetic patients despite controlling traditional risk factors such as hypertension, smoking, hypercholesterolemia, and physical inactivity suggests that prothrombotic state may be the more important factor in these patients. Moreover, a subdued response to standard antiplatelet agents reported in diabetic patients may also explain heightened cardiovascular risk. Therefore, a better understanding of the pathophysiology of atherothrombosis in DM patients may improve the benefits of current pharmacological therapy (e.g., antiplatelet therapy) by maximizing its clinical efficacy and safety.
The purpose of this article is to review the current status of biologic knowledge on platelet hyperreactivity, to evaluate the clinical benefits and limitations of currently available antiplatelet agents, and to suggest future directions to overcome these limitations by new agents and treatment strategies.
PROTHROMBOTIC STATE IN DIABETES MELLITUS
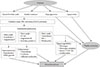
Diabetes is a "prothrombotic state" often characterized by hyperglycemia, oxidative stress, endothelial dysfunction, platelet activation, hypercoagulability with dysfunctional coagulation pathways and fibrinolysis, and inflammation (Fig. 1) [4,5,6]. Platelets activation and aggregation at the site of plaque rupture is pivotal for the subsequent atherothrombotic complications of arterial systems. Platelets in DM patients appear to be hyperreactive with intensified adhesion, activation, and aggregation [6]. Moreover, platelets influence diverse endothelial and inflammatory responses during the initiation and progression of atherosclerosis.
Several mechanisms are suggested to explain the platelet dysfunction in DM patients [6]: hyperglycemia enhances platelet aggregation by increasing P-selectin expression, by osmotic effects, by activating protein kinase C, and by glycating platelet surface proteins with a consequent decrease in membrane fluidity. In addition, insulin resistance or deficient action in diabetic patients are associated with impaired responses to antithrombotic molecules (such as prostacyclin and nitric oxide) and insulin receptor substrate-dependent effects are associated with an increase in the intraplatelet calcium concentration and subsequent enhanced degranulation. Metabolic conditions associated with DM (i.e., obesity, dyslipidemia, and systemic inflammation) may also have a role in this process. Finally, upregulation of glycoprotein (GP) IIb/IIIa expression and P2Y12 signaling, increased platelet turnover, and excessive oxidative stress further contribute to the platelet dysfunction in these patients. Furthermore, different cutoff points of high platelet reactivity (HPR) for adverse events in DM patients compared with the overall population following percutaneous coronary intervention (PCI) have been reported [7,8]. Therefore, diabetic subjects need a personalized antiplatelet therapy strategy to reduce atherothrombotic events associated with hyperreactive platelets.
CLINICAL EVIDENCES OF ANTIPLATELET REGIMEN IN DIABETES MELLITUS
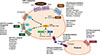
There are multiple targets for antiplatelet therapy (Fig. 2) [9,10]. Atherosclerotic plaque rupture, erosion or fissure exposes the subendothelial matrix and release prothrombotic factors during CVD or PCIs. These processes result in localized platelet adhesion and subsequent platelet activation results in the release of soluble agonists such as thromboxane A2 (TXA2), adenosine diphosphate (ADP), and generation of thrombin on the activated platelet surface by coagulation. TXA2 is produced from arachidonic acid and binds to TX receptors; ADP is secreted from dense granules and binds to platelet P2Y1 and P2Y12 receptors. These agonists, through an autocrine and paracrine fashion, produce sustained activation of GPIIb/IIIa receptors leading to stable platelet-rich thrombus generation. Platelet activation also results in the exposure of phosphatidyl serine, providing binding sites for coagulation factors. The coagulation process results in the generation of thrombin and subsequent platelet-fibrin clot formation. Endogenous phosphodiesterase (PDE) activity affects intraplatelet cyclic adenosine monophosphate (cAMP) levels and modulates platelet function. Finally, isoprostanes derived from membrane arachidonic acid through peroxidation have been shown to induce platelet aggregation by activating the receptor for TXA2.
Importantly, the relative contribution of each pathway (ADP-platelet, TXA2-platelet, thrombin-platelet, coagulation, and PDE activity) to the development of thrombus formation is unknown at this time and can be different depending on the disease entity and activity. Therefore, determination of the optimal combination of antiplatelet agents remains an elusive goal. Occurrences of recurrent ischemic events and bleeding events during contemporary antiplatelet therapy may be related in part to the nonselective "one-size-fits-all" dosing that ignores the inherent variability in thrombogenecity and antiplatelet responsiveness.
Aspirin
Aspirin selectively and irreversibly acetylates cyclooxygenase-1 (COX-1), thereby blocking platelet TXA2 formation and diminishing platelet aggregation mediated by TXA2 (Fig. 2). This effect is irreversible because platelets are enucleate and unable to resynthesize COX-1. In healthy subjects, even low doses of aspirin (~40 mg daily) cause an almost complete suppression of TXA2 formation and platelet aggregation throughout the entire platelet lifespan [11]. However, aspirin therapy in DM patients has a high prevalence of hyporesponsiveness or "aspirin resistance" [12] leading to concerns regarding its effectiveness in the primary prevention of CVD. Because clinical studies used different assays, agonists, cutoff values, and cohorts, interpretation of the data and generalization in clinical practice may be difficult.
Primary prevention
In patients without prior CVD (primary prevention), indication for antiplatelet therapy remains unclear [13]. In this population, aspirin, the only antithrombotic drug studied in a sufficiently large cohort, shows a statistically significant reduction in the risk for a first MI attack at the expense of increased risk of both gastrointestinal (GI) bleeding and hemorrhagic stroke. However, the clinical benefit of aspirin on MI protection can be different according to concomitant use of standard regimen (e.g., angiotensin-converting enzyme inhibitor and statin). In a recent analysis, the clinical benefit of aspirin was not observed in randomized clinical trials (RCTs) published after 2000 (risk ratio [RR], 0.98; 95% confidence interval [CI], 0.84 to 1.14), in contrast to those published before 2000 (RR, 0.67; 95% CI, 0.56 to 0.81; Pinteraction<0.001) [14]. In the meta-analysis by the Antithrombotic Trialists' (ATT) collaboration, aspirin therapy increased major GI and other extracranial bleeds (defined as "a bleed requiring transfusion or resulting in death") (0.10%/year vs. 0.07%/year; RR, 1.54; 95% CI, 1.30 to 1.82; P<0.0001) compared with placebo [15]. When treated with aspirin, the high-risk population would experience 22 more bleeds per 1,000 persons versus 4 more bleeds per 1,000 persons in the low-risk population [16]. A meta-analysis of 16 placebo-controlled RCTs (n=55,462) showed that treatment with aspirin was associated with an increased risk of hemorrhagic stroke by 1.84-fold (P<0.001) [17]. In absolute terms, one could predict 12 incident cases of hemorrhagic stroke per 10,000 patients during chronic aspirin treatment.
During primary CVD prevention that includes subjects with a low risk of developing atherothrombotic events, it is essential to estimate the individual risk-benefit ratio profile, in this case bleeding and hemorrhagic risk [13]. Cardiovascular risk can increase proportionally across primary prevention in young healthy individuals to high-risk individuals and then to secondary prevention (Fig. 3). Aspirin can be recommended for primary cardiovascular prevention based on a threshold risk level, defined as major cardiovascular events (death, MI, or stroke) ≥2 per 100 person-years [13]. An "uncertainty area" at risk levels between 1 and 2 per 100 patient-years should be considered in which the decision to prescribe aspirin is left to the physician's discretion and to the patient's preferences. Moreover, recently the Food and Drug Administration (FDA) has reviewed the available data and does not believe that the current evidence supports the general use of aspirin for primary prevention of a heart attack or stroke. The FDA suggested that it should not be routinely used for primary prevention due to serious risks including increased risk of cerebral and GI bleedings.
Three RCTs conducted specifically in patients with diabetes and six RCTs in which DM patients were subgroups (1% to 22%) failed to show definitive results on the benefit of aspirin in primary CVD prevention (Table 1). A meta-analysis of these nine RCTs found that aspirin therapy was associated with numeric reductions in CAD events (-9%) and cerebrovascular events (-11%) [18]. Based on the overall negative results of these RCTs, it was considered that standard aspirin therapy may be less effective in patients with diabetes than in individuals without diabetes [13]. As such, the current evidence suggests that diabetes should be considered as a unique high-risk entity.
A position statement by the American Diabetes Association (ADA), the American Heart Association, and the American College of Cardiology Foundation recommended that low-dose aspirin (75 to 162 mg daily) for primary prevention is reasonable for DM adults without a previous history of vascular disease who are at increased CVD risk (10-year CVD risk over 10%) without an increased risk for bleeding. This generally includes men over 50 years of age and women over 60 years of age who also have at least one of the following major risk factors: smoking, hypertension, dyslipidemia, family history of premature CVD, and albuminuria [18]. Furthermore, aspirin is no longer recommended for those at low CVD risk (women under 60 years of age and men under 50 years of age with no major CVD risk factors; 10-year CVD risk under 5%). Clinical judgment should be applied for those at intermediate CVD risk (younger patients with one or more risk factors or older patients with no risk factors; those with a 10-year CVD risk of 5% to 10%) until further research is available.
Secondary prevention
The clinical benefit of aspirin therapy is clearly superior to the risk of major bleeding in the setting of secondary CVD prevention. Aspirin is still the bedrock of antiplatelet therapy for secondary prevention of recurrent ischemic events in patients with atherothrombotic disease, including those with DM [19]. The recommended dose of aspirin for secondary prevention in DM patients with atherosclerotic disease is 75 to 162 mg daily. Low-dose aspirin usage is supported mainly by two large meta-analyses of secondary prevention trials performed by the ATT' collaboration involving 212,000 high-risk patients (with acute or previous vascular disease or some other predisposing condition implying an increased risk of occlusive vascular disease) [15,20]. The results of these meta-analyses showed oral antiplatelet agents, mainly aspirin, to be protective for vascular events in high-risk patients. In particular, the incidence of vascular events was reduced from 22.3% to 18.5% in DM patients (P<0.002) and from 16.4% to 12.8% (P<0.00001) in non-DM patients. Although the overall incidence of vascular events was much higher in DM patients, the benefit of antiplatelet therapy was consistent regardless of DM status [20]. In these trials, low-dose aspirin (75 to 150 mg daily) was found to be at least as effective as higher daily doses, and bleeding complications were reduced with lower doses. The first large-scale RCT comparing high- (300 to 325 mg daily) versus low-dose (75 to 100 mg daily) aspirin therapy was the Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent EveNTs-Optimal Antiplatelet Strategy for InterventionS 7 (CURRENT-OASIS 7) trial that included ACS patients scheduled to undergo early coronary angiography [21,22]. The rate of 30-day ischemic events did not differ between high-dose versus low-dose aspirin. However, a trend toward higher rates of GI bleeds was observed in the high-dose versus low-dose group (0.38% vs. 0.24%, P=0.051).
P2Y12 receptor antagonists
Thienopyridines (ticlopidine, clopidogrel, and prasugrel) are nondirect irreversible antagonists of the P2Y12 receptor. Clopidogrel is currently the most commonly prescribed antiplatelet agent. It has similar efficacy and better safety profile compared to ticlopidine. Clopidogrel is a prodrug and needs two-step hepatic conversion to become an active metabolite (Fig. 4) [23]. Numerous data have demonstrated a close relationship between low response to clopidogrel or "clopidogrel resistance" and atherothrombotic events in high-risk patients with acute coronary syndrome (ACS) or those treated with coronary stenting [24]. Because DM itself is an important determinant for clopidogrel responsiveness, an intensified antiplatelet regimen may reduce the risk of "clopidogrel resistance" and consequently the rate of ischemic event occurrence for secondary prevention. Compared with the standard dose of clopidogrel (300 mg loading or 75 mg daily maintenance), high-dose clopidogrel (600 mg loading or 150 mg daily maintenance) strategy is associated with enhanced platelet inhibition and reduced risk for HPR [25], but the high dose strategy can't efficiently overcome the risk of HPR to ADP.
Prasugrel is a third-generation thienopyridine and a prodrug that requires one-step hepatic conversion to its active metabolite to irreversibly inhibit the P2Y12 receptor (Fig. 4). Prasugrel has a more rapid onset of action than clopidogrel and provides greater platelet inhibition because of a more effective conversion into its active metabolite [24]. The Optimizing Antiplatelet Therapy in Diabetes Mellitus-3 (OPTIMUS-3) trial showed that prasugrel (60 mg loading followed by 10 mg maintenance) achieved significantly greater platelet inhibition compared with double-dose clopidogrel (600 mg loading dose followed by 150 mg maintenance) in CAD patients with DM on long-term aspirin treatment, using multiple pharmacodynamics measures [26].
Ticagrelor is a non-thienopyridine, direct-acting, oral antagonist that binds reversibly to the P2Y12 receptor (Fig. 4). The major metabolite of ticagrelor (AR C124910XX), formed by metabolism via the hepatic cytochrome (CYP) 3A4, is as potent as the parent compound ticagrelor. Compared with clopidogrel, ticagrelor results in faster and greater platelet inhibition, with less patient-to-patient variation. In a crossover study including ACS patients with DM (n=30), ticagrelor treatment (90 mg twice daily for 15 days) showed significantly greater platelet inhibition compared with prasugrel treatment (10 mg daily for 15 days) (45.2 vs. 80.8 P2Y12 reaction units measured by the Verify Now P2Y12 assay; P=0.001) [27].
Primary prevention
Currently, the ADA recommends the use of clopidogrel in very high-risk DM patients or as an alternative therapy in patients intolerant to aspirin [19]. However, the use of dual antiplatelet therapy (DAPT) with aspirin and clopidogrel in DM patients without overt atherosclerotic disease has not been supported by clinical evidence.
The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial has compared clopidogrel (75 mg daily)+low-dose aspirin (75 to 162 mg daily) to placebo+low-dose aspirin in high-risk patients (n=15,603, a median follow-up of 28 months), for primary as well as secondary prevention [28]. In this trial, the rates of major vascular events were not significantly different between the two groups. There was a trend towards higher risk of severe bleeding in the primary prevention group compared with the secondary prevention group. In the primary prevention subgroup with multiple risk factors (n=3,284, 80.8% were diabetics), the rate of the primary endpoint was 6.6% with clopidogrel+aspirin versus 5.5% with placebo+aspirin (P=0.20). In addition, there was a significant increase in cardiovascular death (3.9% in the clopidogrel group vs. 2.2% in the placebo group, P=0.01) and also all-cause mortality in the clopidogrel group (5.4% vs. 3.8, P=0.04) [29]. In addition, the rates of severe and moderate bleedings were 2.0% and 2.2% in the clopidogrel group, and 1.2% and 1.4% in the placebo group, respectively (P=0.07 and P=0.08). There is evidence to suggest that atherosclerotic plaques in DM patients are characterized by increased neovascularization of the vasa vasorum [30], which may be associated with a higher risk of intraplaque hemorrhage with consequent rupture or thrombosis.
Secondary prevention
(1) Clopidogrel versus aspirin
The Clopidogrel versus Aspirin in Patients at Risk of Ischemic Events (CAPRIE) trial evaluated the clinical benefits of clopidogrel (75 mg daily) versus high-dose aspirin (325 mg daily) in a secondary prevention population including approximately 20% of DM patients (n=3,866) [31]. The results showed a significantly lower annual risk of the composite endpoint (vascular death, MI, or ischemic stroke) with clopidogrel (5.32% vs. 5.83%, P=0.043). The benefit of clopidogrel therapy was higher in the DM subgroup (15.6% vs. 17.7%, P=0.042), leading to 21 vascular events prevented for every 1,000 DM patients treated [32].
(2) Clopidogrel+aspirin versus placebo+aspirin
Among patients with documented prior MI, ischemic stroke, or symptomatic peripheral artery disease in the CHARISMA trial (n=9,478, ~30% were diabetics), the rate of cardiovascular death, MI, or stroke was significantly lower in the clopidogrel group than in the placebo group: 7.3% vs. 8.8% (hazard ratio [HR], 0.83; 95% CI, 0.72 to 0.96; P=0.01) [33]; this benefit was more prominent in patients with prior MI or ischemic stroke than symptomatic peripheral artery disease (HR, 0.774 vs. 0.780 vs. 0.869). There was no significant difference in the rate of severe bleeding (1.7% vs. 1.5%; HR, 1.12; 95% CI, 0.81 to 1.53; P=0.50). Therefore, the antiplatelet effect of DAPT may reduce the risk of ischemic event occurrence in selected patients with overt CVD outside ACS.
CAD patients with ACS or treated with PCI have a high thrombotic risk and a low responsiveness to aspirin, especially in DM patients; hence, the rationale for combination antiplatelet strategies involves pathways different from TXA2. Multiple placebo-controlled RCTs have demonstrated the clinical benefits of adjunctive clopidogrel combined with aspirin therapy during short- and long-term follow-up (Table 2) [34,35,36]. Although ischemic events were reduced with clopidogrel both in nondiabetic and diabetic patients, diabetic patients showed higher rate of ischemic event occurrences and diminished benefit from adjunctive clopidogrel compared with nondiabetic patients. Thus, patients with DM receive fewer benefits from standard-dose clopidogrel in the setting of ACS or PCI. Intensified inhibition of the platelet ADP-P2Y12 pathway may guarantee more clinical benefits in these patients.
(3) High-dose clopidogrel+aspirin versus standard-dose clopidogrel+aspirin
The CURRENT-OASIS 7 trial evaluated the 30-day clinical benefit of high-dose (600 mg loading followed by 150 mg daily for 1 week) versus standard-dose clopidogrel (300 mg loading followed by 75 mg daily) in ACS patients [21,22]; the subgroup undergoing PCI suggested a clinical benefit in the high-dose group, with a significant reduction in the ischemic event rate (3.9% vs. 4.5%, P=0.039) and stent thrombosis (0.7% vs. 1.3%, P=0.0001) at the expense of major bleeding (1.6% vs. 1.1%, P=0.009) (Fig. 5). Reduction in ischemic events by high-dose clopidogrel was similar in patients with versus without DM (Table 2).
(4) Prasugrel+aspirin versus clopidogrel+aspirin
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel-Thrombolysis in Myocardial Infarction 38 (TRITON-TIMI 38) evaluated the efficacy and safety of prasugrel (60 mg loading followed by 10 mg daily maintenance) versus standard-dose clopidogrel (300 mg loading followed by 75 mg daily maintenance) in moderate- to high-risk ACS patients undergoing PCI (n=13,608) [37]. Prasugrel treatment showed a significant reduction in the rates of the primary endpoint (cardiovascular death, nonfatal MI, or nonfatal stroke) compared with clopidogrel treatment over a follow-up period of 15 months (9.9% vs. 12.1%; HR, 0.81; P<0.001), as well as a reduction in the rates of stent thrombosis at the expense of an increased risk of major bleeding in the prasugrel group (Table 2, Fig. 5). No net clinical benefit was observed in elderly patients (≥75 years) and in those weighing <60 kg; a net harm was found in patients with a history of stroke or transient ischemic attack. Compared with non-DM patients, DM patients tended to have a greater reduction in ischemic events (30% vs. 14% reduction; Pinteraction=0.09) without an observed increase in major bleeding rates [38]. This benefit was consistent in patients with (14.3% vs. 22.2%; HR, 0.63; P=0.009) and without insulin treatment (11.5% vs. 15.3%; HR, 0.74; P=0.009). Importantly, although major bleeding was higher in DM patients, there was no difference in major bleeding among DM patients treated with prasugrel versus clopidogrel (2.6% vs. 2.5%; HR, 1.06; P=0.81).
(5) Ticagrelor+aspirin versus clopidogrel+aspirin
The PLATelet inhibition and patient Outcomes (PLATO) trial explored the issue of whether upstream administration of ticagrelor improves clinical outcome versus clopidogrel in patients with ST-segment-elevation myocardial infarction (STEMI) or NSTE-ACS (n=18,624) [39]. The PLATO trial demonstrated that ticagrelor, when compared to clopidogrel, reduced ischemic events in ACS patients irrespective of diabetes status and glycemic control, without an increase in major bleeding. In PLATO, reduction of the primary endpoint at 1 year (composite of cardiovascular death, MI, or stroke) by ticagrelor was significant and similar both in patients with and without DM (12% vs. 17% relative risk reduction; Pinteraction=0.49). Among patients planned for an invasive strategy, the benefit of ticagrelor was also observed irrespective of diabetic status (HR, 0.88 in diabetic patients and 0.83 in nondiabetic patients; Pinteraction= 0.72). Importantly, ticagrelor was not associated with an increase in protocol-defined major bleeding, although a higher rate of major bleeding not related to coronary artery bypass grafting was observed (4.5% vs. 3.8%; HR, 1.19; P=0.03).
(6) Cangrelor
Cangrelor is an intravenous, direct, reversible, and potent P2Y12 inhibitor. Platelet inhibition is immediate after bolus infusion, the antiplatelet effect is maintained during a continuous infusion and platelet function is restored within 1 hour after discontinuation. Among clopidogrel-naïve CAD patients on aspirin therapy, cangrelor provided dose-dependent blockade of platelet P2Y12 receptors measured by platelet function testing, without different effects according to diabetic status [40]. In a patient-level pooled analysis from the three randomized A Clinical Trial Comparing Cangrelor to Clopidogrel Standard Therapy (CHAMPION) trials including PCI patients (n=24,910) [41], cangrelor versus control (clopidogrel or placebo) significantly reduced the risks of primary endpoint (composite of death, MI, ischemia-driven revascularization, or stent thrombosis at 48 hours) (3.8% vs. 4.7%; OR, 0.81; 95% CI, 0.71 to 0.91; P=0.0007), without differences in GUSTO severe or life-threatening bleeding at 48 hours (0.2% in both groups): no specific interaction between diabetic status and cangrelor efficacy was found.
Adjunctive use of third agent
Despite improved clinical efficacy of DAPT with COX-1 inhibitor aspirin and a potent P2Y12 receptor inhibitor such as prasugrel and ticagrelor, recurrent ischemic event (~10%/year) and increased risk of bleeding episode observed in a significant percentage of ACS [37,39] suggests a ceiling effect of the current DAPT in attenuating ischemic events and some atherothrombotic events are mediated by other pathway(s). Several drugs with different mechanisms have been proposed for use as an adjunctive treatment to DAPT. Agents that have the potential of this "triple therapy" strategies include GP IIb/IIIa inhibitor, PDE inhibitor, protease-activated receptor-1 (PAR-1) antagonists, and new oral anticoagulants (Fig. 4).
Glycoprotein IIb/IIIa inhibitor
GP IIb/IIIa inhibitors are intravenous antiplatelet agents showing the highest benefit in high-risk patients with ACS undergoing PCI, but questionable efficacy in low- to moderate-risk ACS patients or in those treated with a conservative approach [42].
The benefit of GP IIb/III inhibitor pretreatment during clopidogrel therapy appears more pronounced in high-risk ACS patients, including those with DM undergoing PCI. The Intracoronary Stenting and Antithrombotic Regimen: Is Abciximab a Superior Way to Eliminate Elevated Thrombotic Risk in Diabetics (ISAR-SWEET) trial (n=701) did not show beneficial effects of abciximab over placebo (8.3% vs. 8.6%) on the risk of 1-year death and MI in diabetic patients undergoing elective PCI after high-dose clopidogrel (600 mg) pretreatment (HR, 0.97; 95% CI, 0.58 to 1.62; P=0.91) [43]. The Intracoronary Stenting and Antithrombotic: Regimen Rapid Early Action for Coronary Treatment 2 (ISAR-REACT 2) trial (n=2,022) demonstrated a significant reduction of 30-day major adverse cardiac event (MACE) with the use of abciximab versus placebo in patients with NSTE-ACS undergoing PCI on top of 600-mg clopidogrel loading (8.9% vs. 11.9%; OR, 0.75; 95% CI, 0.58 to 0.97; P=0.03) [44], which benefit was restricted to patients with elevated troponin levels (OR, 0.71; 95% CI, 0.54 to 0.95; P=0.02) and was observed across all subgroups, including diabetic patients. The Early Glycoprotein IIb/IIIa Inhibition in Non-ST-Segment Elevation Acute Coronary Syndrome (EARLY-ACS) trial compared strategy of early (~24 hours before PCI) routine administration with delayed provisional administration of eptifibatide (n=9,492) [45], in which the rate of 30-day death or MI did not differ (11.2% vs. 12.3%; OR, 0.89; 95% CI, 0.79 to 1.01; P=0.08) with the expense of higher risks of bleeding and red-cell transfusion in the early eptifibatide group; absolute reduction of MACE at 96 hours with early eptifibatide treatment was more pronounced in patients with versus without DM (2.1% vs. 0.8%). Additionally, a meta-analysis evaluating the effects of GP IIb/IIIa inhibitors in the setting of primary PCI for STEMI suggested a decrease in mortality, but not in re-infarction in diabetic patients [46].
In the era of potent P2Y12 inhibitor, it may be questionable whether diabetic patients may achieve further clinical benefit from the routine use of GP IIb/IIIa inhibitor in ACS patients. In TRITON, the benefit of prasugrel over clopidogrel on primary ischemic endpoint was irrespective of GP IIb/IIIa inhibitor during the index hospitalization (11% and 16% risk reduction in patients with and without GP IIb/IIIa inhibitor) [37]. In the subset of the PLATO trial planned for an invasive strategy, the clinical benefit of ischemic endpoints with ticagrelor versus clopidogrel was numerically lower in patients receiving GP IIb/IIIa inhibitor (10% and 19% risk reduction in patients with and without GP IIb/IIIa inhibitor) (Pinteraction=0.37) [47]. A major concern with routine use of GP IIb/IIIa inhibitor on top of potent P2Y12 inhibitor is the increase in the risk of serious bleeding. The provisional injection of GP IIb/IIIa inhibitor (intravenous or intracoronary) with short-term infusion (~6 hours) in the selected cases as a bridging strategy (e.g., angiographic evidence of massive thrombus, slow or no-reflow, or a thrombotic complication) may be optimal strategy to maximize clinical efficacy and safety during potent P2Y12 inhibitor therapy.
Phosphodiesterase inhibitor
Mammalian phosphodiesterases (PDEs) are the important targets for pharmacologic intervention in the treatment of a number of diseases such as erectile dysfunction, pulmonary hypertension, intermittent claudication, and chronic pulmonary obstructive disease [48]. Therefore, many new PDE inhibitors are being developed for treatment of these disorders. The superfamily of PDEs is comprised of 11 families of enzymes, and individual isozymes modulate distinct regulatory pathways in different cells. For example, PDE5 isozymes are found in platelets, vascular smooth muscle and endothelial cells, with observed high expression in corpus cavernosum and lung. PDE2, PDE3, and PDE5 isozymes are accountable for the majority of platelet PDE activity (>90%) [49]. In platelets, cyclic adenosine 3',5'-monophosphate (cAMP) is hydrolysed by PDE3 and PDE2, whereas cyclic guanosine 3',5'-monophosphate is hydrolysed by PDE5 and PDE2. Dual mechanism with increased production of cAMP (by clopidogrel) and decreased degradation of cAMP (by PDE inhibitor) synergistically enhances the level of intraplatelet vasodilator-stimulated phosphoprotein-phosphorylation and thus stabilize platelet activation. "Triple therapy" with adjunctive PDE3 inhibitor cilostazol to DAPT (aspirin+clopidogrel) significantly enhances platelet inhibition compared with double-dose clopidogrel in high-risk patients (e.g., HPR, AMI, DM, and so on) [50]. On the other hand, other PDE inhibitors pentoxifylline (nonselective) and dipyridamole (PDE5) did not enhance ADP-mediated platelet inhibition similar to cilostazol [51]. The latter finding may be mainly related to different effect of PDE inhibitors on intraplatelet cAMP levels. Contrary to cilostazol, pentoxifylline and dipyridamole have weak effect on intraplatelet cAMP levels, which may be associated with their low PDE3 selectivity.
Cilostazol is a dual inhibitor of PDE3 and adenosine reuptake that may have an important role in reducing ischemic events associated with CAD [50-53]. Cilostazol is a widely used selective and reversible PDE3 inhibitor, which is highly expressed in myocardial and vascular smooth muscle cells (VSMCs) and platelets. It also inhibits adenosine reuptake into erythrocytes, endothelial cells, muscle cells, and platelets, thereby increasing interstitial and circulatory adenosine levels at clinically relevant concentrations (~3 µmol/L). Adenosine activates G-protein-coupled adenosine receptors, possesses a wide range of biological activities and influences cell survival through pre- and post-conditioning processes in experimental studies. In platelets and VSMCs, the interaction of adenosine with Gs-coupled adenosine A2 receptors results in increased intracellular cAMP. Thus, cilostazol can increase the production and also inhibit the breakdown of cAMP in platelets and VSMCs. The unique feature of cilostazol may contribute to the observed efficacy profile of cilostazol in platelet reactivity and atheroma progression among DM patients. For example, the Diabetic Atherosclerosis Prevention by Cilostazol (DAPC) trial compared prevention by cilostazol (100 to 200 mg daily) versus aspirin (81 to 100 mg daily) of progression in carotid intima-media thickness in type 2 diabetic patients during a 2-year observation period [54]. The regression in maximum left and right common carotid artery intima-media thickness was significantly greater with cilostazol compared with aspirin (-0.088±0.260 mm vs. 0.059±0.275 mm, P<0.001; -0.042± 0.274 mm vs. 0.045±0.216 mm, P=0.003). In the Adjunctive Cilostazol versus double-dose ClopidogrEL in Diabetes Mellitus (ACCEL-DM) trial, adjunctive cilostazol to DAPT showed the greater inhibition of platelet aggregation and the lower prevalence of HPR than double-dose clopidogrel in type 2 diabetic patients undergoing PCI [50]. More interestingly, compared with clopidogrel (75 mg daily) on top of aspirin, adjunctive cilostazol (100 mg twice daily) to aspirin showed the similar inhibition of ADP-induced platelet aggregation [52,53]. In addition, the cilostazol treatment achieved the lower level of platelet function after the stimuli with collagen and arachidonic acid compared with the clopidogrel treatment, which implicates the unique character of antiplatelet effect by cilostazol.
The benefit of this triple therapy strategy has been mostly observed in PCI-treated patients, mainly as a reduction in the rates of target lesion revascularization and even in stent thrombosis [55,56,57]. In a recent meta-analysis, adjunctive cilostazol reduced the risk of angiographic restenosis irrespective of stent type (51% and 37% relative reduction after bare-metal stent and drug-eluting stent, respectively) and decreased numerically the risk of stent thrombosis by 43% (95% CI, 0.41 to 1.67), without the increase of major bleeding (OR, 1.00) [55]. The clinical efficacy of cilostazol in ischemic events may be more prominent in the setting of ACS. In a Chinese clinical trial including ACS patients (n=1,212), triple antiplatelet therapy with the addition of 6-month cilostazol after successful PCI was associated with a significantly lower incidence of the primary endpoint (composite of cardiac death, nonfatal MI, stroke, or target vessel revascularization at 1 year) (10.3% vs. 15.1%; HR, 0.65; 95% CI, 0.41 to 0.91; P=0.011), and no differences in the risks of TIMI major or minor bleeding were found (0.2% vs. 0.2%) [56]. Of note, the DM subgroup showed a more pronounced benefit with triple therapy (53% reduction) (Fig. 5). However, the use of cilostazol is limited by the high frequency of side effects (e.g., headache, palpitations, and GI disturbances) and increased risk of withdrawal.
PAR-1 inhibitor
Thrombin is the serine protease enzyme linked between plasmatic and cellular components of the thrombotic process and it plays a crucial role in the platelet activation and coagulation cascade [58]. Platelet PAR-1 and PAR-4 account for the thrombin-mediated signaling in platelets. PAR-1 mediates platelet responses at subnanomolar concentrations of thrombin, whereas PAR-4 mediates platelet activation at higher thrombin concentrations. Activation of either one is sufficient to trigger platelet secretion and aggregation, whereas PAR-1 is likely to be the most important receptor. In addition to platelet-mediated effects and fibrin polymerization during clot generation, thrombin exerts diverse effects on various cells. The PAR-1 receptor is present in platelets, endothelial cells, VSMCs, mononuclear cells, fibroblasts, and cells of atherosclerotic plaque, suggesting a major role in tissue response to injury, angiogenesis, inflammation, and thrombosis. In addition to its role during initial thrombus generation by stimulating platelet aggregation, thrombin that is produced in large quantities following thrombus generation may stimulate the secretion of platelet-derived growth factor and induce angiogenesis. The latter response to thrombin may contribute to vascular remodeling and also restenosis.
Five PAR-1 antagonists have been developed, of which only one drug (vorapaxar) has been investigated in phase III clinical trial and approved for treatment in ACS patients. Vorapaxar is an oral competitive PAR-1 antagonist that blocks thrombin-mediated platelet activation without interfering with thrombin-mediated cleavage of fibrinogen [58]. It is rapidly absorbed (peak level in 60 to 90 minutes), has high bioavailability and a half-life of approximately 311 hours. Following promising findings from early phase clinical investigations, vorapaxar was tested in two large-scale, phase III clinical trials.
In the Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome (TRACER) trial (n= 12,944) [59], the clinical efficacy and safety of vorapaxar (40 mg loading dose and a 2.5 mg daily maintenance dose) versus placebo in addition to standard antiplatelet therapy (96% on aspirin and 91.8% on clopidogrel) was evaluated in patients with NSTE-ACS. Vorapaxar versus placebo treatment was associated with a significant decrease in the composite of cardiovascular death, MI, or stroke at 2 years (14.7% vs.16.4%; HR, 0.89; 95% CI, 0.81 to 0.98; P=0.02), at the expense of the increase in the rate of GUSTO moderate or severe bleeding (7.2% vs. 5.2%; HR, 1.35; 95% CI, 1.16 to 1.58; P<0.001), and a 3-fold increase in intracranial bleeding (1.1% vs. 0.2%, P<0.001). The excess prevalence of intracranial hemorrhage in patients with a history of stroke led to an unplanned safety review, which recommended early termination of this trial.
In the Trial to Assess the Effects of Vorapaxar in Preventing Heart Attack and Stroke in Patients With Atherosclerosis-TIMI 50 (TRA 2P-TIMI 50) trial (n=26,449) [60], secondary prevention by adjunctive vorapaxar (2.5 mg daily) versus placebo in addition to standard-of-care therapy (58% on DAPT) was assessed among patients with known atherothrombotic disease (a history of MI, ischemic stroke, or peripheral arterial disease). Vorapaxar significantly reduced the primary endpoint (composite of cardiovascular death, MI, or stroke) compared with placebo at 30-month follow-up (9.3% vs. 10.5%; HR, 0.87; 95% CI, 0.80 to 0.94; P<0.001), which was largely driven by a 17% reduction in the MI risk. GUSTO moderate or severe bleeding occurred in 4.2% of patients who received vorapaxar and 2.5% of those who received placebo (HR, 1.66; 95% CI, 1.43 to 1.93; P<0.001) and intracranial bleeding was a twofold increase by vorapaxar (1.0% vs. 0.5%, P<0.001). Contrary to patients with a history of stroke, patients with previous MI (n=17,779) treated with vorapaxar exhibited a reduction in the primary endpoint at 3-year compared with placebo (8.1% vs. 9.7%; HR, 0.80; 95% CI, 0.72 to 0.89; P<0.0001) [61]. Despite an overall increase in bleeding complications, intracranial bleeding were not significantly higher in the vorapaxar versus placebo group (0.6% vs. 0.4%, P=0.076). The clinical benefit by vorapaxar was even more pronounced after exclusion of elderly patients (>75-year old), individuals with a history of stroke, and those with a low body weight (<60 kg). In diabetic patients with a prior MI (n=3,623) [62], vorapaxar significantly reduced the primary endpoint (11.4% vs.14.3%; HR, 0.73; 95% CI, 0.60 to 0.89; P=0.002) with a number needed to treat to avoid 1 major cardiovascular event of 29. The incidence of GUSTO moderate or severe bleeding was increased with vorapaxar in DM patients (4.4% vs. 2.6%; HR, 1.60; 95% CI, 1.07 to 2.40). However, net clinical outcome integrating these two endpoints (efficacy and safety) was improved with vorapaxar (HR, 0.79; 95% CI, 0.67 to 0.93).
Based on the subanalysis, the FDA approved clinical use of vorapaxar (2.5 mg daily) in addition to standard-of-care therapy (aspirin, clopidogrel, or both) among patients with a history of MI or with peripheral arterial disease. Vorapaxar is contraindicated in patients with a history of stroke, transient ischemic attack, or intracranial hemorrhage, and in those with active pathological bleeding.
RESISTANCE TO ANTIPLATELET AGENT IN DIABETIC PATIENTS
Numerous data have demonstrated a close relationship between HPR or "antiplatelet resistance" and atherothrombotic events in high-risk patients (e.g., PCI-treated patients with ACS or DM) [24]. In "laboratory resistant" patients, antiplatelet drug fails to block its specific platelet target (e.g., aspirin against COX-1 enzyme and clopidogrel against P2Y12 receptor) and it is only meaningful when "laboratory resistance" is translated "treatment failure" (the recurrence of ischemic events despite treatment).
Prevalence of aspirin resistance is widely variable across the studies that may be due to differences in platelet function testing used, definition of resistance, aspirin dose, and patient cohort. When COX-1-dependent tests (by determination of serum/urine thromboxane and assays with arachidonic acid as agonist) are used, aspirin resistance is a rare phenomenon (<5% of patients) [63,64] and the main cause of this aspirin resistance is poor compliance. However, when COX-1-independent tests assays are used, prevalence of aspirin resistance appears higher. The Aspirin-Induced Platelet Effect (ASPECT) study demonstrated that aspirin inhibited platelet aggregation stimulated by agonists other than arachidonic acid in a dose-dependent manner among stable CAD patients [63]; significant effects were observed for collagen- and shear-induced aggregation and 11-dehydrothromboxane B2 production. The latter finding may be due to effects of aspirin beyond inhibition of its primary target COX-1 by acetylation and was termed a non-COX-1 effect.
The attenuated antiplatelet effect of aspirin therapy in DM patients can be explained by various mechanisms such as reduced drug bioavailability, accelerated platelet turnover, and glycosylation of platelet membrane proteins [6]. When platelet turnover is heightened, an increased proportion of immature platelets capable of protein synthesis are released from the bone marrow and can be identified as a marker of accelerated thrombopoiesis. In a post hoc analysis of ASPECT, greater platelet reactivity and a higher prevalence of aspirin resistance were present in the patients with DM [65]. Aspirin doses of >81 mg daily (162 to 325 mg daily) were associated with similar rates of resistance and platelet function in patients with and without DM. A higher aspirin dosing strategy than 81 mg daily in DM patients may be associated with enhanced platelet inhibition (mainly by COX-1-dependent methods) and possibly better protection against atherothrombotic event. Elevated TXA2 synthesis may be related with increased platelet turnover in DM patients; the introduction of newly generated platelets not exposed to aspirin into the systemic circulation continues to generate TXA2, which may activate thromboxane and prostaglandin endoperoxide (TP) receptor. TP receptor activation has led to interest in developing TP receptor blockers [6].
In a post hoc analysis of ASPECT, a higher aspirin dose (162 to 325 mg daily) than 81 mg daily did not decrease the level of ADP-mediated platelet function and closure time in PFA-100 collagen/epinephrine assay among stable CAD patients with DM [65]. In aspirin-treated patients presenting for angiographic evaluation of CAD (n=562), both serum thromboxane B2 >3.1 ng/mL and PFA-100 collagen-ADP closure time <65 seconds (OR, 3.5; 95% CI, 1.2 to 10.4; P=0.027) were associated with MACEs at 2-year follow-up [64]. This finding suggests that multiple mechanisms, including but not confined to inadequate inhibition of COX-1, are responsible for poor clinical outcomes in aspirin-treated patients. The addition of other pathway blockade (e.g., P2Y12 inhibitor) can be plausible strategy to overcome the combined risk of aspirin resistance in DM patients. Since enhanced inhibition of platelet activation by combination regimen can increased the risk of serious bleeding, the potency of antiplatelet therapy must be determined on the risk profile of the patient cohort. In the primary prevention subgroup with multiple risk factors from CHARISMA (n=3,284, 80.8% were diabetics) [29], clopidogrel versus placebo on top of aspirin did not decrease the rate of the primary endpoint (6.6% vs. 5.5%, P=0.20) and increased the risk of severe bleeding (2.0% vs.1.2%, P=0.07).
DAPT with clopidogrel and aspirin is the standard antiplatelet regimen in high-risk DM patients (e.g., ACS or PCI). However, a substantial portion of DM patients suffers from recurrent cardiovascular events. The prevalence of "clopidogrel resistance" varies considerably and is related to differences in definitions, type of test used, clopidogrel dose, and cohort character [24]. Genetic, cellular, and clinical mechanisms have been associated with inadequate responsiveness to clopidogrel. The presence of DM is an important clinical factor that contributes to "clopidogrel resistance." Numerous mechanisms have been suggested to explain the inadequate clopidogrel response observed in DM patients: low bioavailability of clopidogrel, lack of response to insulin in platelets, alterations in calcium metabolism, upregulation of P2Y12 receptor signaling, increased exposure to ADP, and increased platelet turnover [6]. Several antiplatelet treatment strategies have been developed to optimize platelet inhibition: (1) dose modification of clopidogrel; (2) use of potent P2Y12 inhibitor agents; and (3) addition of a third antiplatelet drug (triple therapy) (e.g., cilostazol, PAR-1 inhibitor) [9]. There is an accompanying increased risk of bleeding with more potent platelet inhibition. It could be an important issue in the future trials whether a therapeutic window exists for antiplatelet strategy to simultaneously limit thrombotic and bleeding events.
CONCLUSIONS
Diabetes itself is a hypercoagulable state and hyperreactive platelets in DM patients remarkably contribute to the increased risk of ischemic events occurrence. Furthermore, DM patients have shown low response to commonly used antiplatelet regimen (aspirin and clopidogrel). Understanding mechanism of "treatment failure" in DM patients during antiplatelet therapy may enable more reasonable approaches to maximize clinical efficacy and safety. Because the role of aspirin in primary prevention among DM patients still remains questionable, upcoming results from ongoing aspirin trials in primary prevention and clinical evidences from other treatment strategies (e.g. statin, P2Y12 antagonist, and polypill) are warranted. For secondary prevention in high-risk DM patients (e.g. ACS), the development of more potent or new combination antithrombotic strategies may control the enhanced hypercoagulable state in diverse pathways and therefore improve clinical outcomes. Large-scale randomized trials specifically designed to evaluate these new antithrombotic strategies in DM patients are warranted to determine their efficacy and safety.




 XML Download
XML Download