

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The number of patients with diabetes continues to increase drastically worldwide, and the number of affected individuals in the world is currently estimated at 386.7 million in the 2014 report of the International Diabetes Federation [1]; thus, accounting for a prevalence of 8.3% in the global population aged between 20 and 80 years old or 1 in 12 individuals, with this number estimated to increase to 591.9 million by 2035. Similarly, obesity is also shown to continue to increase worldwide, with the number of overweight/obese individuals reported to have increased in 2013 to 2.1 billion in the Global Burden of Disease study [2]. Of note, under a background of insulin resistance, obesity is shown to induce diabetes, dyslipidemia and hypertension or the metabolic syndrome; thus, contributing to a high incidence of cardiovascular disease among individuals with obesity [34]. Given that individuals with the metabolic syndrome are thought to be at high risk of type 2 diabetes and that the continued increase in number of patients with diabetes represents a considerable challenge, there remains a critically urgent need to elucidate the causes of obesity, insulin resistance and diabetes and their complications as well as to establish definitive/curative preventive/therapeutic modalities for these diseases.
Identified at our laboratories in 2003, the adiponectin receptors (AdipoRs) AdipoR1 and AdipoR2 are shown to be membrane proteins implicated in glucose/lipid metabolism that become activated by adipocyte-derived adiponectin and are evolutionarily conserved in living beings from plants to humans. Once activated by adipocyte-derived adiponectin, AdipoR1 and AdipoR2 activate AMP-activated protein kinase (AMPK) and peroxisome proliferator-activated receptor (PPAR), respectively, and promote glucose/lipid metabolism; thus, exerting their anti-diabetic properties. Therefore, elucidation of the mechanisms through which AdipoRs become activated is thought to lead to improvements in the metabolic syndrome as well as anti-diabetic approaches and generate clues for the development of novel anti-diabetic therapeutic modalities. Of note, we identified adiponectin receptor agonist (AdipoRon), an AdipoR-activating small-molecule compound, in 2013, and went on to establish the crystal structures of the AdipoRs using X-ray crystallography in 2015, which revealed that the AdipoRs have novel structures, with their seven-transmembrane helices each enclosing a large cavity that contains a zinc ion, being conformationally distinct from those of G-protein coupled receptors (GPCRs). Thus, we were the first to establish the AdipoRs, long predicted to be seven-transmembrane proteins, as membrane proteins whose structure has an opposite orientation in the membrane from those of GPCRs, which are widely known as seven-transmembrane proteins whose N- and C-termini are located extracellularly and intracellularly, respectively. It is expected that these conformational findings will contribute to the development of safe and highly efficacious AdipoR-targeted, the drugs to extend lifespan as well as to their optimization.
ANTI-DIABETIC ACTION OF ADIPONECTIN
While obesity was long known to induce hyperglycemia, dyslipidemia and hypertension, i.e., the metabolic syndrome, through a background of insulin resistance, the mechanisms whereby obesity induces insulin resistance remained largely unclear. Currently, obesity as an underlying cause of the metabolic syndrome is assumed to occur primarily due to enlargement of adipocytes. In addition to their known role in storing excess energy in the form of triglycerides, adipocytes have come to be recognized as an organ secreting various "adipokines" or signaling molecules such as leptin, tumor necrosis factor α (TNF-α), resistin, and free fatty acids (FFAs) [56789]. It has also become clear that some of those secreted by the enlarged adipocytes including TNF-α, resistin, and FFAs interfere with insulin signaling in skeletal muscle; thus, inducing insulin resistance.
Conversely, adiponectin has come to be recognized as an adipokine improving insulin sensitivity. Adiponectin is an adipocyte-derived, secreting protein with a molecular weight of about 30 kDa, consisting of a signal peptide at the N-terminus, a collagenous domain, and a globular domain at the C-terminus [10111213].
In our experiments, high-fat diet-loading was shown to induce adipocyte enlargement as well as to aggravate insulin resistance in a mouse model of type 2 diabetes, with marked decreases in blood adiponectin levels [14]. Thus, in an attempt to clarify the implications of decreased adiponectin in the presence of enlarged adipocytes, the mice with high-fat diet-induced obesity was given adiponectin supplementation, which led to improvements in insulin resistance and dyslipidemia, suggesting that adiponectin has insulin-sensitizing properties [15]. To complement this findings, Fruebis et al. [16] found that globular forms of adiponectin promote fatty acid burning, and Berg et al. [17] and Combs et al. [18] also reported that adiponectin improves insulin sensitivity in the liver and inhibit gluconeogenesis, thus reducing blood glucose levels.
Furthermore, adiponectin-knockout mice were shown to be associated with insulin resistance, impaired glucose tolerance, dyslipidemia and hypertension; thus, presenting with the metabolic syndrome, suggesting that adiponectin deficiency has a key role to play in the pathogenesis of the metabolic syndrome [19202122].
It was thus suggested that decreases in adiponectin associated with obesity account at least in part for impaired glucose tolerance, dyslipidemia and hypertension, which are primarily responsible for the metabolic syndrome.
It is also shown that adiponectin increases the expression of acyl CoA oxidase (ACO) implicated in FFA burning in skeletal muscle as well as of uncoupling proteins (UCPs) involved in energy consumption. Given that these genes are PPAR-α targeted genes, experiments were conducted to examine PPAR-α expression in mouse models of altered insulin sensitivity, which demonstrated that adiponectin administration led to increases in the expression of PPAR-α [15], as well as in endogenous PPAR-α ligand activity [23].
Again, adiponectin is also shown to promote FFA burning in C2C12 cells, an in vitro model of skeletal muscle [24]. In this regard, non-transcription-mediated pathways are reported to be available to mediate fatty acid burning through phosphorylation of AMPK [25]. AMPK is shown to become activated with physical activity and is thus considered a key molecule involved in the supply of energy required for physical activity by promoting non-insulin-dependent glucose uptake and fatty acid burning.
Interestingly, adiponectin has been shown to activate AMPK. Thus, it is suggested that increased FFA burning, glucose uptake and utilization promoted by adiponectin in skeletal muscle as well as glucose lowering in vivo with acute adiponectin administration may be accounted for at least in part by AMPK activation [24]. In this regard, quite apart from our experiments, Tomas and colleagues [26] also reported that globular adiponectin activates AMPK.
CLONING OF ADIPORs
Given that decreased plasma adiponectin levels directly increase the risk factors for diabetes and dyslipidemia and also directly affect vasculature [2027] and is thus responsible for the metabolic syndrome, and associated macroangiopathy, it is suggested that adiponectin effects-enhancing treatments may constitute definitive modalities for diabetes and macroangiopathy. Thus, as it was thought critical to the development of such definitive therapeutic modalities to elucidate the mechanisms of action of adiponectin, our efforts focused on identifying AdipoRs.
By exploring adiponectin-specific binding, we were the first in the world to identify AdipoR1 and AdipoR2 [28]. It is shown that AdipoR1 and AdipoR2 share high homology (66.7% amino acid) in the sequence that is conserved from yeasts to humans. Of note, the yeast AdipoR1 homologue YOL002c is shown to play a key role in metabolic pathways such as fatty acid oxidation [29]. AdipoR1 is relatively ubiquitously expressed in the entire body but abundantly expressed in skeletal muscle, while AdipoR2 is particularly abundantly expressed in the liver. Predicted as novel seven-transmembrane receptors with an N-terminus located intracellularly and a C-terminus located extracellularly, the AdipoRs are characterized as having a topology opposite to those of GPCRs ever reported in the literature. In this regard, we showed in experiments using siRNA that AdipoR1 and AdipoR2 are required for adiponectin binding to the cell membrane surface in cultured cells [29], and went on to generate AdipoR1 or AdipoR2 knockout mice, and showed that no adiponectin binding occurs and adiponectin-associated metabolic actions disappear in AdipoR1/R2 double knockout mice, thus demonstrating that the AdipoRs represent key receptors on metabolic pathways [30].
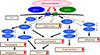
Again, given that AdipoR1/R2 double knockout mice exhibit insulin resistance and impaired glucose tolerance, increased inflammation and oxidative stress leading to increased gluconeogenesis and decreased glucose uptake in key metabolic organs such as liver, skeletal muscle and adipose tissue are suggested as the mechanisms responsible for these conditions (Fig. 1) [30].
PHYSIOLOGICAL AND PATHOPHYSIOLOGICAL ROLES OF THE ADIPORs IN VARIOUS TISSUES
We have shown that both AdipoR1 and AdipoR2 are decreased in animal models of obesity and type 2 diabetes and that this decreased AdipoR expression is responsible in part for the onset of diabetes. We have also shown that upregulation of AdipoR1 expression in the liver leads to AMPK activation, while upregulation of AdipoR2 expression activates PPAR-α, promotes fatty acid burning and energy consumption, and mediates anti-inflammatory and anti-oxidative stress effects, thereby improving impaired glucose tolerance [30].
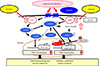
We have also demonstrated that adiponectin/AdipoR1 signaling upregulates metabolism and exercise endurance by increasing mitochondrial volume and function in a similar fashion to physical activity [31]. The expression of PPAR-α coactivator-1 (PGC-1α) [32] was shown to be decreased to 25% of its normal level in newly engineered skeletal muscle-specific AdipoR1-knockout mice, with decreases seen in mitochondrial volume and function, type 1 fiber, leading to decreased exercise endurance as well as impaired glucose tolerance and insulin resistance at the organism level. Again, we have also shown through experiments using C2C12 myotubes or Xenopus laevis oocytes that adiponectin increases intracellular calcium concentrations and activates AMPK/longevity gene sirtuin 1 (SIRT1) [33] via AdipoR1; thus, constituting exercise-mimicking signaling. It was also found that increased intracellular calcium concentrations contribute to increased expression of PGC-1α, while activation of AMPK/SIRT1 contributes to activation of PGC-1α, suggesting that, together, these pathways constitute a dual mode of PGC-1α regulation (Fig. 2) [31].
THE ADIPOR AGONIST ADIPORON AND ITS ANTI-DIABETIC EFFICACY
Altering the metabolic capacity of an organism through activation of adiponectin/AdipoR signaling can contribute greatly to normalizing its metabolic environment. Thus, the development of adiponectin- or AdipoR-activating drugs or AdipoR agonists is eagerly awaited in that they have high potential as "exercise-mimetics" providing similar effects to those of exercise; thus, opening the way not only for definitive treatment of the metabolic syndrome, type 2 diabetes or atherosclerosis but for effective treatment of these diseases even in those who have difficulty in exercising due to an internal disease or locomotor disorders.
In screening for candidate compounds using the chemical library at the University of Tokyo's Open Innovation Center for Drug Discovery, we have succeeded in identifying a small-molecule AdipoR-activating compound (AdipoRon) [34]. AdipoRon was shown to bind to AdipoR1 and AdipoR2 directly and activate AMPK in skeletal muscle thus increasing mitochondrial function; it was also shown to improve metabolic capacity in the liver, skeletal muscle, and adipose tissue via the AdipoRs as well as to exert anti-diabetic properties.
LIFE-PROLONGING EFFECTS OF THE ADIPOR AGONIST ADIPORON
Of the mechanisms through which adiponectin improves insulin resistance via AdipoR1, we identified one which involves AdipoR1-mediated AMPK activation by adiponectin in the liver and skeletal muscle (Figs. 1 and 2) [24283031]. We also clarified that the adiponectin/AdipoR1 pathway increases the nicotinamide adenine dinucleotide (NAD+)/NADH ratio in skeletal muscle thereby activating the longevity gene SIRT1 [31].
Again, it was also found that adiponectin upregulates via AdipoR2 the expression of ACO involved in fatty acid burning as well as UCP involved in energy consumption [2830]. Given that the ACO and UCP promoters are shown to contain a PPAR-responsive element, we went on to examine ACO and UCP for their endogenous PPAR-α ligand activity and found that their endogenous PPAR-α ligand activity is increased and PPAR-α expression is also increased by adiponectin [28]. We also found that the adiponectin/AdipoR2 pathway increases the expression of catalase and superoxide dismutase (SOD); thus, alleviating oxidative stress at the organ level [30].
Caloric restriction is widely known to prolong lifespan [35]. It is recently suggested that AMPK, mechanistic target of rapamycin (mTOR) [36] and SIRT account in part for the mechanisms through which caloric restriction leads to prolonged lifespan. In this regard, overexpression of AMPK α subunit is shown to lead to prolonged lifespan in Caenorhabditis elegans [3738]. It is also assumed that AMPK blocks mTOR signaling and inhibits protein synthesis thereby inhibiting cancer cell proliferation and neoangiogenesis. Numerous studies show that mTOR signaling inhibition leads to prolonged lifespan in yeasts, nematodes and drosophila. It is also shown that the use of the mTOR inhibitor rapamycin prolongs lifespan in mice [30].
It is well recognized that obesity leads to increased tissue oxidative stress, which adversely affects aging and lifespan. Conversely, it is known that overexpression of the oxidative stress-relieving genes catalase and SOD leads to prolongation of lifespan [394041].
Against this background and given that adiponectin/AdipoR signaling activates the AMPK/SIRT1 pathway and positively regulates catalase and SOD; thus, relieving oxidative stress in various tissues, we examined lifespan in AdipoR-knockout mice, on the assumption that lifespan may be shortened in these mice, and demonstrated that AdipoR1 or AdipoR2-knockout mice are associated with a shorter lifespan than high-fat diet-fed wild-type mice and that AdipoR1/AdipoR2-double knockout mice have the shortest lifespan [34].
Again, the AdipoR agonist AdipoRon was shown to improve metabolic capacity in the liver, skeletal muscle and adipose tissue and to exert anti-diabetic properties. Additionally, while high-fat diet loading results in a shortened lifespan in mouse models of obesity and type 2 diabetes, it has been shown that administration of AdipoRon leads to normalization of obesity-shortened lifespan despite high-fat diet loading in these mouse models [34].
STRUCTURES OF HUMAN ADIPORs
The AdipoRs were predicted to be seven-transmembrane receptors each with an opposite topology to that of a GPCR whose N- and C-termini were known to be located intracellularly and extracellularly, respectively. While the mechanisms through which GPCRs become activated began to be unraveled through structure analysis of GPCR complexes formed with trimeric G proteins downstream, the structures of the AdipoRs remained largely unknown. The structural information about AdipoR1 and AdipoR2, if available, should be very important for developing and optimizing AdipoR agonists. Thus, we focused attention on elucidating the conformational structures of the AdipoRs by means of X-ray crystallography and their functions based on their structures.
Only recently have we engineered anti-AdipoR antibodies capable of recognizing the conformation of the AdipoRs and succeeded in crystalizing the AdipoR/anti-AdipoR antibody (Fv fragment) complexes by using lipidic mesophases [42]. Based on the crystals obtained, we have determined the crystal structures of AdipoR1 and AdipoR2 at 2.9 and 2.4 Å, respectively [43].
AdipoR1 and AdipoR2 were shown to be quite similar in structure, each composed of an N-terminus intracellular domain, a short intracellular helix, seven-transmembrane helices, and a C-terminus extracellular domain. It was also confirmed that the anti-AdipoR antibody used in crystalizing the AdipoRs recognized the N-terminus intracellular domain.
Thus, we went on to search the available conformation-dependent protein library for proteins analogous in structure to the AdipoRs but no such protein was found in the library. It was also found that the AdipoR seven-transmembrane domain whose C-terminus is located extracellularly has an opposite orientation in the cell membrane from that of any bacterial rhodopsin or GPCR and that the AdipoRs lack the proline-induced kinks that characterize the structures of GPCRs; thus, suggesting that the AdipoR1 and AdipoR2 represent a novel class of receptor structures.
We also found that within the seven-transmembrane domain of AdipoR1 and AdipoR2 contains a zinc binding side located at a distance of about 4 Å from the inner cell membrane and that the zinc ion was coordinated at a distance of 2.1 to 2.6 Å by three His amino acid residues; that a water molecule was located between the zinc ion and the carboxylate side-chain of Asp; and that the three His/Asp residues are conserved within the mammalian AdipoR homologues.
We further examined the zinc ion coordination for correlation with AMPK activation by altering the zinc ion coordination-related amino acid to Ala and demonstrated that zinc-binding in AdipoR1 may not be directly required for AMPK activation but may be effective for stabilization of the AdipoR1 structure. In contrast, we found that zinc-binding in AdipoR2 may be directly related not only to the stabilization of the AdipoR2 structure but to the AdipoR2 signaling pathways, thus leading to our hypothesis that AdipoR2 may have zinc ion-dependent hydrolytic activity.
The seven-transmembrane domain of both AdipoR1 and AdipoR2 was shown to have a cavity with a zinc-binding site, which contains unidentified extra electron densities. It was thus suggested that these electron densities may represent potential substrates for AdipoR hydrolytic activity or their products. For development of best-in-class AdipoR agonists, optimization of AdipoRon based on 3D structure of AdipoRon-AdipoR complex should be most important.
CONCLUSIONS
Recent structural analysis of AdipoR1 and AdipoR2 revealed that they are structurally different and functionally distinct from GPCRs thus representing a novel class of receptors. Elucidating the structures of the AdipoRs may not only lead to a better understanding of signaling pathways associated with the AdipoRs as seven-transmembrane receptors but have important implications for optimization of the AdipoR agonist "AdipoRon" as a first-in-class drug toward becoming a best-in-class drug. Thus, current data on the crystal structures of the AdipoRs [43] may be expected to accelerate the development and optimization of AdipoR-targeted small-molecule AdipoR-activating compounds such as "AdipoRon" [34]. In the years to come, small-molecule AdipoR-activating compounds are expected to be further refined as AdipoR-activating agents and developed as effective therapeutic modalities for the treatment of patients with the metabolic syndrome and diabetes, which contribute to healthy life expectancy in these individuals (Fig. 3).

 XML Download
XML Download