

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
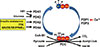
At present, metabolic diseases including diabetes, non-alcoholic fatty liver disease, cardiovascular disease, and cancer are the most life-threatening diseases. The imbalance of glucose and free fatty acid (FFA) homeostasis primarily causes metabolic diseases. Although this perspective has been challenged [1], the glucose-FFA cycle, the Randle cycle, is a paradigm for interpreting of metabolic regulation and treating of metabolic diseases such as diabetes and cancers [2]. According to the Randle cycle, β-oxidation increases acetyl-CoA (Ac-CoA), nicotinamide adenine dinucleotide reduced (NADH), and flavin adenine dinucleotide reduced (FADH2) in mitochondria. The increased [Ac-CoA]/[CoA-SH] and [NADH]/[NAD] ratio allosterically inhibits the pyruvate dehydrogenase complex (PDC) activity [3] and activates the pyruvate dehydrogenase kinases (PDKs), resulting in indirectly reducing the PDC activity [4]. PDC, a key regulatory enzyme complex in glucose oxidation, mediates the oxidative decarboxylation of pyruvate, which converts pyruvate to Ac-CoA and CO2 and reduces NAD+ to NADH. Therefore the ratio of β-oxidation activity and PDC activity controls the glucose-FFA metabolism, which involves the cellular fuel selection between glucose and FFA depend on the physiological status [5]. In fed state, PDC is highly active to oxidize the glucose to generate energy or to convert the glucose to fat to store the energy in peripheral tissues and liver. In starved state, PDC in peripheral tissues is inactive to inhibit the glucose oxidation and to spare the three carbon compounds (lactate, alanine, and pyruvate), which are substrates for gluconeogenesis, leading to maintain the euglycemia in fasting condition [6]. In addition to starved states, the dysregulation of glucose metabolism by the lowering of PDC activity causes various metabolic diseases including diabetes [78], cancer [910], and sepsis (Fig. 1) [11]. In insulin resistance or obesity, the reduced PDC activity in peripheral tissues and hepatocytes spares the gluconeogenic substrates and increases the hepatic glucose production, which induces the hyperglycemia, resulting in promoting the progression into diabetes. Furthermore, hypoxia-inducible factor 1 (Hif-1)-mediated induction of PDKs also inhibits PDC activity, which decreases the reactive oxygen species (ROS) in the tumor cells, resulting in induction of cancer cell proliferation and metastasis [910]. It therefore is critically important to adequately maintain PDC to prevent the occurrence of metabolic diseases.
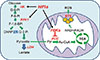
PDC consists of three components such as pyruvate dehydrogenase (E1), dihydrolipoyl acetyltransferase (E2), and dihydrolipoyl dehydrogenase (E3) (Fig. 2). In addition to allosteric regulation by end products including Ac-CoA and NADH, PDC is also regulated by covalent modification such as phosphorylation in a long-term manner [12]. PDKs phosphorylate the pyruvate dehydrogenase (PDH)-E1α and inactivates PDC (Fig. 2) [12]. The phosphorylated PDH-E1α can be reactivated via the dephosphorylation by the pyruvate dehydrogenase phosphatases (PDPs). According to reports in mammals, there are four PDK isoenzymes (PDK1 to 4) [13] and two PDPs (PDP1 and PDP2) [614]. Although the phosphorylation-dephosphorylation cycle of PDC is an important regulation mechanism, the effect of PDPs on the PDC regulation and the expressional regulation of PDPs have received less attention compared to those of PDKs [15]. Huang et al. [15] reported that PDP1, which is a Ca2+-sensitive isoform, is expressed in rat heart, brain, and testis and that PDP2, which is a Ca2+-insensitive isoform, is abundantly expressed in rat kidney, liver, heart, and brain. They also observed that only PDP2 is down-regulated by starvation and the diabetic state in those tissues. Compared to PDP, the effects of PDKs on PDC regulation and the expressional regulation of PDKs have gained more attention during the last two decades.
The expression of PDK isoenzymes is regulated by a physiological condition in tissue-specific manners [161718]. Among the four PDK isoenzymes, the effect of PDK4 on the regulation of PDC activity has been extensively studied. Recently, the transcriptional regulation of PDK2 and PDK4 by Forkhead box factor 1 (Foxo1) and nuclear hormonal receptors (peroxisome proliferate activation receptor [PPAR], estrogen receptor [ER], glucocorticoid receptor [GR], estrogen-related receptor [ERR], and thyroid receptor [TR]) has been summarized (Fig. 2) [19]. However, PDK1 and PDK3 have received less attention than PDK4 and PDK2 because those isoenzymes are less abundant than PDK4 and PDK2 and are expressed in a highly tissue-specific manner [16]. However, Kim et al. [9] have reported that Hif-1α-induced PDK1 causes the Warberg effect, which reprograms the glucose metabolism from oxidative glycolysis to aerobic glycolysis (Fig. 3), in cancer cells. After the publication of this report, studies regarding the expressional regulation of PDK1 and its effect on the regulation of PDC activity have dramatically increased [20212223].
In this review, we summarize the recent advances of the roles of specific PDK isoenzymes on the induction of metabolic diseases, such as obesity, insulin resistance and diabetes, and cancers. We also summarize the effects of PDK inhibition on the prevention of metabolic diseases using pharmacological inhibitors and genetically modified animals.
ROLE OF PYRUVATE DEHYDROGENASE KINASES ON THE REGULATION OF PDC ACTIVITY IN METABOLIC SYNDROMES
Pyruvate dehydrogenase kinase 1
Since it has been revealed that PDK1 is responsible for the Warberg effect in cancer cells, a lot of studies focuses on the effect of PDK1 on the cancer metabolism and how cancer cells can survive in hypoxic condition. Hif-1, an oxygen-sensing transcription factor, consists of Hif-1α and Hif-1β [24]. In the normoxic condition, Hif-1α is lowered by an ubiquitin-dependent proteasomal degradation induced by the von Hippel-Lindau protein, resulting in the prevention of heterodimerization with Hif-1β [25]. In the hypoxic condition, Hif-1α is stabilized and forms the active dimer with its partner, resulting in the up-regulation of target genes involved in metabolism [26], angiogenesis [27], and cell proliferation [28]. In cancer cells, Hif-1α induces the Warberg effect by the induction of glycolytic enzymes due to an abnormal stabilization of Hif-1α (Fig. 3) [29]. PDK1 has been reported to be one of the target genes of Hif-1 in hypoxia [923], pulmonary hypertension [30], and cancer [31]. Since PDC and electron transport system of mitochondria are major sources of cellular ROS, the inactivation of PDC by the up-regulation of PDK1 reduces the production of ROS from the both pathways, which saves cancer cells from apoptosis [23]. The traditional strategies of cancer therapy are ineffective in several solid tumors associated with the induction of Hif-1α and its target genes, which reduce ROS production. Oppositely, the pharmacologic inhibition of Hif-1α, resulting in decreasing the PDK1 expression, increases the oxygen consumption in cancer cells and inhibits cancer cell proliferation due to activation of PDC [20]. In addition to stabilize Hif-1α by hypoxia, McFate et al. [28] found that glycolytic metabolites such as lactate and pyruvate, which is increased by the lowering of PDC activity by PDK1, also stabilize Hif-1α via a hypoxia-independent manner and establish a positive loop of Hif-1α stabilization, suggesting that obesity or insulin resistance could increase the development of cancers.
Pyruvate dehydrogenase kinase 2
PDK2 is most abundantly and constitutively expressed in all tissues [16]. However, its expression is slightly increased in several tissues such as skeletal muscles, adipose tissues, and liver by starvation or insulin resistance [1732]. In PDK2 knockout (PDK2-KO) mice, the blood glucose level of the PDK2-KO mice is slightly but significantly lower than that of wild type (WT) mice in the postprandial period [33]. However, there is no difference in overnight fasting blood glucose levels as well as PDC activity in several tissues between the PDK2-KO mice and WT mice on a chow diet. These results suggest that PDK2 involves the regulation of PDC activity, whereas the activity of the other kinases is low, such as occurs in the fed state because PDK4 is up-regulated and inhibits the PDC activity in the peripheral tissues during the starved state. However, it has recently been observed that PDK2 deficiency affects the whole-body fat metabolism in mice after a high fat diet, which ameliorates the insulin resistance and hyperglycemia induced by high fat diet (unpublished data).
Additionally, it has been suggested that PDK2 also involves in the proliferation of cancer cells [28]. Since most vascular diseases are caused by uncontrolled proliferation of vascular smooth muscle cell (VSMC), which is similar to cancer cell proliferation, metabolic changes by PDKs could involve the development of vascular diseases. Indeed, dichloroacetate (DCA), a well-known PDK inhibitor as a pyruvate analogue, inhibits the proliferation of VSMCs, leading to prevention of vascular restenosis induced by arterial injury [34]. DCA decreases the mitochondrial membrane potential and the cellular ATP level, which primarily inhibits PDK2 activity in the VSMCs because of DCA. Furthermore the possibility that PDK2 involves in the regulation of the cancer metabolism has been reported. Byun et al. found that retinoic acid-related orphan receptor α (RORα) alters the glucose and glutamine metabolism in hepatic carcinoma cell lines, such as HepG2 and Hep3B [35]. Adenoviral over-expression of RORα and SR1078, a pharmacological RORα activator, inhibits PDK2 expression in hepatoma cell lines via p21 up-regulation and switches the aerobic glycolysis to TCA-mediated oxidative phosphorylation (OxPhos), which results in increases in ROS production and the [NADP]/[NADPH] ratio, leading to inhibited cell proliferation of hepatocarcinomas.
Pyruvate dehydrogenase kinase 3
Thus far, PDK3 has received less attention than other PDK isoenzymes because it has been reported that PDK3 is, for the most part, expressed in the testis, whereas its expression level is extremely low in other tissues [16]. Although PDK3 exhibits the highest specific activity among the PDK isoenzymes, the physiological role in the regulation of PDC activity is little known [163637]. Recently, PDK3 is up-regulated by Hif-1 in cancer cell lines in hypoxic conditions and in stem cells [3839]. Over-expression of PDK3 leads to inducing drug resistance and to deteriorating the prognosis of colon cancer [40]. To date, there is no report that PDK3 is involved in diabetes or obesity.
Pyruvate dehydrogenase kinase 4
PDK4 is the most attractive kinase among the PDK isoenzymes because its expression is dramatically changed by the physiological conditions [1241]. PDK4 is remarkably induced in several peripheral tissues, including skeletal muscle, heart, mammary gland, adipose tissue, and kidney in starved or diabetic mammals and humans [171842]. However, PDK4 is slightly increased in the livers of animals while PDK2 is primarily increased in the livers of starved and diabetic animals [1742], indicating that PDK4 is the main PDK in peripheral tissues and PDK2 is the main PDK in liver to regulate the PDC. The up-regulation of PDK4 could determine the essential fuel between lipids and carbohydrates in peripheral tissues [5]. The reduced PDC activity in the peripheral tissues by the induction of PDK4 spares the three carbon compounds (lactate, pyruvate, and alanine) for hepatic gluconeogenesis to maintain euglycemia during starvation but deteriorates hyperglycemia in diabetic animals. The PDK4-KO mice exhibited hypoglycemia during starvation compared with WT mice fed chow [42]. Furthermore, PDK4-KO mice had lower blood glucose levels and were observed to be more insulin sensitive than WT after consuming a high-fat diet [43]. A reduced supply of gluconeogenic substrate from the peripheral tissues to the liver is responsible for this phenomenon [4243]. Furthermore, the hepatic fat level of PDK4-KO mice is less than that of WT mice after consuming a high saturated fat diet [44]. The results obtained from PDK4-KO mice suggest that PDK4 induction is associated with metabolic diseases, including hyperglycemia, insulin resistance, and hepatic steatosis. Recently, Liu et al. [45] observed that PDK4 also increases cancer cell proliferation because the stabilization of cAMP-response element-binding protein by interaction with PDK4 strongly increases the mammalian target of rapamycin complex 1 activity, which is a major regulator of tumor development. Furthermore, hematopoietic stem cells could maintain their stem cell properties by aerobic glycolysis similarly to cancer cells because of the increased PDK4 expression in a Hif-1α-dependent manner [46].
Which mechanism is responsible for the regulation of PDK4 gene expression has been extensively studied. The Foxo has been proposed to be one of the primary regulators for up-regulation of the PDK4 gene in response to the fed/fasting cycle [47]. In the fed state, insulin completely represses the PDK4 gene by Foxo phosphorylation and Akt activation. During fasting and insulin resistance states, Foxo binds to the insulin response elements on the human PDK4 gene, which induces PDK4 gene expression. Jeong et al. [19] summarized that ERRs, TRs, and fasting-responsive factors, including glucocorticoid, PPARα, liver X receptor, PPARγ coactivator 1α, and free fatty acids, are responsible for the up-regulation of the PDK4 gene.
EFFECTS OF THE SMALL MOLECULAR PDK INHIBITORS ON METABOLIC DISEASES
Up-regulation of PDKs plays a pivotal role in the development of various metabolic diseases. Therefore, PDKs are attractive targets for the development of medications for treating metabolic diseases, especially type 2 diabetes, obesity, hepatic cirrhosis, lactic acidosis, and cancers. The development of small pharmacochemical(s), which target the PDKs, is an attractive approach for treating metabolic diseases induced by dysregulation of glucose metabolism.
Several decades ago, DCA has been known to decrease hyperglycemia in diabetic rodents and induce hypoglycemia in starved animals [4849]. Additionally, DCA has long been used for treating lactic acidosis [50] as well as ketoacidosis [51]. The activation of PDC activity by DCA contributes to acutely reduce blood lactate levels [48]. Although DCA inhibits the uptake of ketone bodies in peripheral tissues, it also inhibits ketogenesis in hepatic cells, leading to an increase in the re-esterification of free fatty acids from peripheral tissues [51]. Because the fundamental mechanism of the Warberg effect in cancer cells has been elucidated [931], DCA has shed new light on the treatment of cancer therapy. DCA has been proven to be effective in a wide spectrum of cancers, including colon, breast, glioblastoma, and oral squamous cell carcinoma [52535455]. In cancer, DCA shifts the glucose metabolism from aerobic glycolysis to glucose oxidation, which increases the OxPhos of mitochondria as well as the ROS production. ROS modulate cell proliferation and increase apoptosis of cancer cells. In addition to the direct effect of DCA on the metabolic changes, DCA treatment prevents the angiogenesis induced by Hif-1α in solid tumors [56], which restricts the supply of fuel resources into solid tumors. Despite the therapeutic and economic benefits of DCA, it has been used in limited situations, such as life-threating lactic acidosis induced by PDH-E1α deficiency or sepsis for the short term because of its toxicities, including neuropathy and hepatic tumorigenesis, poor pharmacokinetics, and low potency and selectivity. Therefore, attempts to develop new medications, which improve the demerits of DCA, have been made for the last two decades.
Medication targets pyruvate dehydrogenase kinases
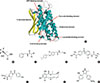
PDC activity is regulated by PDKs and PDPs in a short-term and long-term manner. In the long-term manner, the amount of protein in PDKs is regulated by transcriptional and translational levels due to the physiological conditions. It may not be a suitable target for the development of new agents to target the long-term regulatory mechanism because of the complexities of hormonal regulation in vivo. Therefore, most medication targets are focused on the short-term regulation of PDKs' activity. PDKs should dimerize as homodimers or heterodimers to interact with their substrate, a lipoyl domain of the E2 component of PDC. Based on studies concerning crystal structure, mechanisms of action and effector regulation, sites of binding of effectors, and interaction with other components, three domains of PDKs are important for regulating PDK activity: pyruvate-binding domain (N-terminal regulatory domain), lipoamide-binding domain, and nucleotide-binding domain (C-terminal catalytic domain) (Fig. 4A) [41]. These domains might be good targets for the development of medications.
N-terminal regulatory domain
The N-terminal of PDK consists of the regulatory domain and the lipoamide-binding domain. In the regulatory domain, several allosteric regulators could be bound and regulate the PDK activity in a short-term manner: allosteric activators such as acetyl-CoA and NADH, and allosteric inhibitors such as pyruvate, coenzyme A (CoA-SH), and NAD+. In the last half century, DCA has been extensively studied as a pyruvate analogue. Its low potency and toxicity limit its usage for the short-term period. Thus far, several effective compounds, which are derived from DCA, are successful for inhibiting PDK activity; (R)-trifluoro-2-hydroxy-2-methylpropionic acid (Fig. 4B) and its anilide-derivatives improve the potency by 20- to 40-fold [57]. Administering these compounds orally into diabetic Zucker rats significantly increases the PDC activity in skeletal muscle, kidney, heart, and liver [57]. Halogenated acetophenones, another DCA derivative, also inhibit PDK activity in vitro; however, their potency is not comparable to DCA [58]. Recently, N-phenyl dichloroacetamide (Fig. 4C) and its derivatives have been developed as an anticancer agent that significantly improves the potency compared to DCA (IC50 >1,000 µm vs. 4.76 µm; DCA vs. N-phenyl dichloroacetamide) in an in vitro kinase assay, it induces cancer cell apoptosis, and it has a low toxicity in mice (LD50=1.1 g/kg) [59]. These results suggested that DCA and its analogues might be a good scaffold structure for developing a new medication against PDK, although DCA has a simple structure and is hard to develop derivatives.
In addition to pyruvate, CoA-SH, which is another allosteric inhibitor, also binds to its specific binding site on the regulatory domain of PDKs [60]. A CoA-SH analogue, Pfz3 (Fig. 4D), has been developed via a high-throughput screening system. It's mechanisms of action on PDKs have been studied but still remain uncertain [60]. Recently, phenylbutyrate has been identified to bind in this pocket as well [61]. In this report, phenylbutyrate significantly reduces the lactate concentration in the primary cultured fibroblasts obtained from patients with PDC deficiency and prevents lactic acidosis induced by partial hepatorectomy in mice. Furthermore, phenylbutyrate inhibits the kinase activity of PDK1, PDK2, and PDK3 but not PDK4, indicating that it acts in a kinase-specific manner [62], which suggests that CoA-SH binding site might be a candidate target of kinase-specific medication development.
Lipoamide-binding domain
In the N-terminal of PDKs, there is a lipoamide-binding site(s) that allows PDKs to bind to the PDC-E2 subunit for their activity. Without binding, PDKs could not phosphorylate the PDC-E1α subunit. This domain is also an important potentiated target for developing a PDK inhibitor. AZD7545 (Fig. 4E), its derivative (compound K), and Nov3r (Fig. 4F) target this domain [63]. These Inhibitors have different inhibition kinetics among the PDK isoenzymes [6364], significantly activate PDC activity, and significantly lower blood glucose levels in Zucker diabetic rats.
Nucleotide (ATP)-binding domain
Because ATP is required for the transfer of the phosphoryl group to the PDC-E1α subunit, ATP should bind to the PDKs. The ATP binding site locates the C-terminal catalytic domain of PDK isoenzymes. Therefore, ATP analogues are potential candidates for developing a PDK inhibitor. Radicicol (Fig. 4G), a heat shock protein 90 (HSP90) inhibitor, binds to the ATP-binding domain of PDKs, leading to direct inhibition of ATP binding to this domain [64]. In contrast to radicicol, M77976 (Fig. 4H) is developed as a PDK4-specific inhibitor [65]. It also binds to the same domain; however, the mechanism of action is different from radicicol. The binding of M77976 induces a large conformational change in the nucleotide-binding domain of PDK4, which leads to inhibition of ATP binding affinity [65].
CONCLUSIONS
The up-regulation of PDK isoenzymes deteriorates hyperglycemia induced by obesity and insulin resistance because of the increased hepatic gluconeogenesis and promotes tumorigenesis, proliferation of cancer cells, and metastasis because of switching glucose metabolism from oxidative glycolysis to aerobic glycolysis, the Warberg effect. In this report, we summarized the effect of PDK isozymes on these processes and the regulation of PDK isoenzymes due to the physiological situations, and we discussed the three potential target domains for the development of PDK inhibitors. Thus far, DCA is the most extensively studied compound; however, it has limited use for therapeutic purposes because of its low potency and high toxicity. Although several compounds have been developed for improving DCA limitation in the last two decades, their efficacy still remain to be tested in vivo. Therefore, the efficacy test and the toxicity analysis of those compounds should be conducted in the near future. If the PDC activity is too high, it will induce ketoacidosis and hypothermia [33]. Therefore, the strategy of a PDK inhibitor should be such that it has high specificity among the PDK isoenzymes.

 XML Download
XML Download