

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Structure and function of endothelium
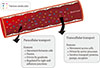
A monolayer of endothelial cells lines the entire circulatory system of the body. This endothelium acts as a barrier which regulates the exchange of hormones, proteins and small molecules between the vascular compartment and the interstitial space [1,2]. The actions of a hormone or nutrient on a target tissue is implicitly dependent upon the ability of these factors to gain access to the target and numerous studies have indicated that hormone and nutrient concentrations in blood differ from those surrounding cells on the tissue side of the blood vessel endothelium [3,4,5,6,7,8,9,10,11]. In this regard, it is our contention that the significance of the endothelium as a regulator of hormone and substrate access to target tissues is often underappreciated. Endothelial permeability can be regulated by two distinct pathways: 1) the transcellular pathway where solutes are actively transported across the endothelium, primarily via caveolae-mediated transcytosis; or 2) the paracellular pathway where solutes passively move through the intercellular space between adjacent endothelial cells (Fig. 1).
TRANSCELLULAR TRANSPORT
The transcellular movement of solutes involves energy-dependent trafficking of vesicles across the endothelium [2]. This often requires their recognition by receptors in caveolae on the luminal surface of the endothelium, may involve vesiculo-vacuolar organelles or occur via transcellular channels [2]. Caveolae, cholesterol- and sphingolipid-rich non-clathrin-coated pits, are abundant in endothelial cells. After ligand binding to receptors in caveolae, dynamin-mediated endocytosis occurs followed by vectorial transport and fusion of the vesicle with basolateral membrane, resulting in the release of contents by exocytosis [2]. As a general concept, transcellular vesicle trafficking is important in the transport of larger macromolecules across the endothelium since the paracellular route is typically capable of restricting passage of solutes larger than 3 nm in radius [12,13].
PARACELLULAR TRANSPORT
The paracellular mode of transport across endothelia depends upon concentration gradients between blood and interstitial fluid. One regulatory mechanism of paracellular movement involves tight junctions (TJs) which are composed of strands of transmembrane and cytosolic proteins that closely control paracellular flux and have been established to be important in regulating hormone transport [4,12,14]. As a component of the cell-cell junction of endothelia, the TJ provides a selective barrier to solute movement between cells [13]. The permselectivity of the TJ barrier is dependent on the variable assemblage of TJ proteins that comprise the complex. Proteins such as occludin, tricellulin, and claudins directly establish the TJ barrier and form the backbone of TJ strands while the cytosolic proteins, such as ZO-1, provide structural support to the TJ complex. The incorporation of specific TJ protein isoforms can enhance TJ barrier function (i.e., make TJs tighter) or form channels to increase TJ permeability (i.e., make TJs leakier) [13]. Importantly, skeletal muscle and heart vasculature have continuous endothelium with TJs between cells (Fig. 2) [15,16]. However, the liver and spleen have a discontinuous endothelium with large holes, which allow rapid equilibration of plasma with the underlying tissue [15,16]. It was suggested that this expedited exposure of the liver to plasma solute changes may partly explain its earlier susceptibility to insulin resistance [17]. Indeed, the precise architecture of TJs varies between different vascular beds, with TJs being either dominant at the apical point of intercellular space or intermingled with adherens junctions (AJs). Therefore, there is no doubt that transendothelial paracellular flux in certain vascular beds is more susceptible to changes in TJ composition and structure. Furthermore, changes in paracellular transport leading to endothelial hyperpermeability is a significant problem in vascular inflammation associated with diabetes, cancer, ischaemia-reperfusion injury, thrombisis, trauma, sepsis, and adult respiratory distress syndrome [18]. Importantly, despite the tremendous interest in adipokines over recent years very few studies have examined the critical step whereby and factors secreted by adipose tissue, or other tissues, must enter the circulation via capillaries or lymph. Paracellular movement is very likely to be a major regulatory step in this process. Indeed, it has been demonstrated that lymph/capillary partitioning of adipokines is dependent upon their molecular size and proposed that this will have important ramifications for their regional and systemic distribution and subsequent physiological effects [19].
It is also important to realize that crosstalk exists between 1) transcellular and paracellular routes of transport across endothelia and 2) TJs and AJs of endothelia. An example of the former is that in caveolin-1 knockout mice, or upon siRNA-mediated reduction of caveolin-1, altered TJ assembly in small capillaries and veins increased paracellular transport of albumin [20,21,22]. In the latter case, it has been shown that vascular endothelial (VE)-cadherin mediated signaling can regulate the expression of TJ proteins and consequently alter barrier function [2].
Indeed, evidence suggests that AJs also play a crucial role in regulating vascular permeability. They are comprised of calcium-dependent VE-cadherin in complex with a range of interacting partners, such as β-catenin and p120-catenin [2] and the expression of VE-cadherin is a specific early developmental marker for endothelial lineage. Deletion of the VE-cadherin gene induces embryonic lethality due to early collapse of the vasculature [23]. Increased vascular permeability occurs when VE-cadherin homophilic binding is inhibited. AJ stability is also critically regulated by phosphorylation [2]. More specifically, phosphorylation of VE-cadherin at catenin binding sites induced internalisation and thus alterations in vascular permeability. Interestingly, recent work demonstrated that phosphorylation of these residues (tyrosine 658 and 685 of VE-cadherin) occurs constitutively in veins but not arteries, correlating with enhanced leakiness in venous vessels [24]. Furthermore, single point mutations in VE-cadherin (Y658F and Y685F) reduce paracellular permeability by blocking internalisation of VE-cadherin [24].
ENDOTHELIAL DYSFUNCTION IN DIABETES
The concept of endothelial dysfunction in diabetes is well established [25]. However, this typically refers to vascular functions independent of transendothelial solute flux. The endothelium is also a dynamic interface that responds to various stimuli and synthesizes and liberates vasoactive molecules such as nitric oxide, prostaglandins and endothelin. Accordingly, vascular complications in diabetes include outcomes occurring in large (atherosclerosis, cardiomyopathy) and small (retinopathy, nephropathy, neuropathy) vessels [26]. We believe that diabetes-induced deleterious alterations in paracellular solute movement also represents a form of endothelial dysfunction which should be fully characterized to establish its physiological significance.
STUDIES ON TRANSENDOTHELIAL TRANSPORT OF HORMONES
Transport of insulin has been perhaps the best studied example. Insulin concentration at the target cell surface has been shown to be very different from that in plasma by approaches including microdialysis [7,8], direct interstitial sampling [9], and lymph measurements [5,6]. Furthermore, early temporal studies have demonstrated that insulin-mediated glucose uptake in muscle lags behind increases in plasma insulin [27], In contrast, when using cultured skeletal muscle cells, the addition of recombinant insulin has been shown to stimulate glucose uptake almost immediately (maximal at~10 minutes) [28]. The time delay seen in vivo has led to the suggestion that reaching the interstitial space is the limiting factor for insulin-mediated glucose uptake [14]. Thus, modification of access to skeletal muscle can have major effects on insulin action and subsequent metabolism [14,29]. Indeed, the efficiency and extent of insulin delivery to the interstitial space can be inhibited physiologically by diet [30]. The mechanism via which insulin crosses the endothelium is at least in part via the paracellular pathway and for more information on this topic readers are referred to recent excellent review articles by Kolka and Bergman [14,31].
Recent years have seen great interest in the role of adiponectin in the pathophysiology of diabetic complications [32,33,34]. Numerous studies have now established that adiponectin can act on various targets to mediate antidiabetic, anti-inflammatory, antiatherosclerotic, and cardioprotective effects [32,34,35]. Thus, enhancing adiponectin action is considered as a therapeutically beneficial strategy. Adiponectin circulates in the blood as three multimeric complexes; low molecular weight (LMW; trimer), medium molecular weight (MMW; hexamer) and high molecular weight (HMW; oligomer), and in obese and diabetic patients reduced circulating levels of adiponectin have been observed. In addition, cleavage of adiponectin by leukocyte derived elastase generates a C-terminal globular fragment of adiponectin which mediates important functional effects. It is likely that delivery of adiponectin to the interstitial space is a major, yet underestimated, determinant of its function. Given the size range of the biologically active forms of adiponectin (16 kDa for globular form and >500 kDa for HMW) there is considerable potential for selective paracellular transport of adiponectin, particularly in different vascular beds. HMW adiponectin is often considered to be the most biologically active and most relevant form with respect to the metabolic syndrome, and we speculate that given the large size of the HMW form, transendothelial movement may prove to be a significant rate-limiting step in its physiological actions (Fig. 3). Indeed, it has been proposed that HMW adiponectin mediates beneficial metabolic effects primarily by acting on liver [34], whereas the globular form is thought to have more potent metabolic effects in skeletal muscle [33]. These observations could be explained by the leaky and moderately tight paracellular barriers present in vascular beds of these respective tissues, although this is likely an oversimplification (Fig. 3). As far as we are aware, no studies have directly examined the paracellular movement of adiponectin in skeletal muscle and liver.
Studies of adiponectin movement across the blood-brain barrier (BBB) have indicated that only LMW and MMW, but not HMW forms were found in cerebralspinal fluid [36,37,38]. Surprisingly, there are only a few studies examining interstitial adiponectin levels. One found that interstitial concentrations of adiponectin in human adipose tissue were ~25-fold lower than plasma [39] while the other found that exercise increased interstitial adiponectin levels, which were around 20% of plasma concentration [40]. We hypothesize that the magnitude of gradient between plasma and interstitial levels of adiponectin in skeletal muscle may be higher due to existence of a tighter endothelial barrier within the muscle vasculature. Indeed, this may be highly significant as it has been shown that hyperinsulinemia differentially affects the compartmental (interstitial and circulating) distribution of the adiponectin complexes in lean and insulin-resistant, obese individuals [41]. We have recently shown that adiponectin moves across cultured endothelial monolayers via a paracellular route and that this movement was reduced by hormonal or physical manipulation of TJs.
TJ COMPOSITION AND PROTEIN EXPRESSION IN DIABETES
Studies examining changes in TJ composition and protein expression in diabetes have not been extensive but have so far yielded numerous consistent observations in the study of diabetic complications. For example, reductions in ZO-1 have been observed in kidney glomeruli, the retina, BBB and intestine of various diabetic models [42,43,44,45,46]. Localization of ZO-1 in glomeruli was also investigated by electron microscopy and found to redistribute from the podocyte membrane to the cytoplasm in the diabetic kidney [47]. Several reports have indicated that both occludin and claudin-5 were reduced in diabetic retina or BBB [44,45,46,48,49]. Hence, expression or localization of TJ proteins appear to be susceptible to a diabetic environment and contribute to complications such as retinopathy and nephropathy. More widespread analysis is now needed, particularly in metabolically active tissues. Knockout mouse models have been generated for various TJ proteins, including occludin and claudin-1, 2, 4, 5, 7, 9, 11, 14, 15, 16, 18, and 19 [50,51,52,53,54,55,56,57,58,59,60,61,62,63]. Little to no information was given regarding their metabolic phenotype, however in one study using Cldn2-/-Cldn15-/- double-knockout mice the animals died from malnutrition related to defective absorption of glucose, amino acids and fats [63]. These models may prove valuable in elucidating the significance of alterations in transendothelial hormone flux in determining metabolic dysfunction in diabetes.
THERAPEUTIC POTENTIAL OF TARGETING ENDOTHELIAL TRANSPORT
Based on the above description of the importance of endothelial transport in diabetes, it is evident that developing therapeutic strategies which manipulate paracellular flux may prove useful. Indeed, a literature review indicates that various agents are already available; such as peptides which bind to integral TJ proteins, siRNA and antisense oligonucleotides targeting TJ proteins, various toxins, lipids, and activators or inhibitors of kinases and phosphatases that regulate junction assembly and function have all been shown to regulate paracellular permeability [64]. Nevertheless, there is an innate risk in therapeutics which globally alter transendothelial permeability and so it will be desirable to elicit changes in a localized or tissue-specific manner, or to targeting specific components of TJs which allow more controlled and selective changes in permeability [65]. One example of a current therapeutic approach targeting TJs is the use of glucocorticoids as a locally applied treatment for diabetic retinopathy. The mechanism of action is thought to involve, at least in part, restoration of barrier function in the retinal vasculature by modifying TJ composition and structure [66]. Therefore, it is attractive to speculate that controlled manipulation of paracellular transport may be applicable to improving metabolic dysfunction in diabetes.
CONCLUSIONS
The endothelial monolayer is an important rate-limiting determinant of hormone and substrate access to target tissues. Previous studies focusing on insulin suggest that impaired delivery of insulin from plasma to interstitial space in obesity may contribute to metabolic insulin resistance and diabetes. Since adiponectin is known to mediate beneficial antidiabetic effects, and the molecular weight of biologically active forms of adiponectin ranges widely, we propose that paracellular transport may be a major determinant of transendothelial adiponectin flux. In particular, the research community has often accepted that fact that the HMW oligomeric form of adiponectin targets liver and not muscle whereas the small globular portion has 'more potency' in skeletal muscle. This was initially attributed to the higher binding affinity of globular fragment of adiponectin to skeletal muscle cell membranes which were thought to express more adiponectin receptor AdipoR1 isoform. However, such data are not consistent and easier access of the smaller globular adiponectin across the relatively tight endothelial monolayer found in skeletal muscle vasculature, in comparison with liver, may be significant. Thus, TJ-mediated, or AJ-mediated, alterations in adiponectin access to tissues from the vasculature are likely to be of important functional significance. Numerous studies in kidney and retina have indicated that expression of various TJ proteins is altered in diabetes; however, the changes in metabolic target tissues of adiponectin and their physiological significance still need to be established. The principal mechanisms responsible for altered TJ composition and structure in diabetes must also be elucidated, with likely mediators including hyperglycemia, hyperinsulinemia, free fatty acids or tumor necrosis factor-α.
Overall, the paracellular permeability of the endothelial barrier is a tightly regulated process which can be dynamically adjusted in response to various mediators. The potential contribution to metabolic dysfunction in diabetes is apparent yet the precise significance has likely been somewhat underestimated. We believe that further investigation of TJ-mediated changes in hormone or substrate flux in diabetes and characterization of their functional significance will shed additional light on the pathophysiology of disease and identify potential therapeutic opportunities.


 XML Download
XML Download