

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
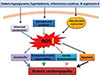
Diabetic cardiomyopathy (DCM) is one of the major cardiac complications in diabetic patients. Several mechanisms responsible for DCM have been proposed [1,2,3]: (1) impaired regulation of intracellular calcium, leading to impaired cardiac contractility; (2) mitochondrial dysfunction, leading to over-production of reactive oxygen species (ROS), reactive nitrogen species (RNS), and eventually cardiac cell death; (3) accumulation of advanced glycated end-products in the heart, leading to extracellular matrix accumulation that in turn results in cardiac diastolic dysfunction and eventually functional failure; (4) abnormal cellular metabolism, leading to accumulation of toxic lipids in the heart; and (5) essential trace metal dyshomeostasis such as zinc and copper. Although these pathogeneses may be primarily caused by hyperglycemia, other pathogenic factors including hyperlipidemia, inflammatory cytokines, and angiotensin system such as angiotensin II also play important roles in the initiation or progression of DCM. These pathogenic factors cause the pathogenesis of DCM probably via different mechanisms, but all these effects are thought related to oxidative stress [1,3,4,5,6], as illustrated in Fig. 1.
ANTIOXIDANT THERAPY AND METALLOTHIONEIN
Oxidati vestress indicates a severe imbalance between ROS and/or RNS generation and their clearance by antioxidant defense systems [3]. Due to low contents of antioxidants in the normal heart compared to other organs, the heart is a highly susceptible organ to oxidative stress and damage [7,8]. Diabetes may up-regulate several antioxidants in the heart as a compensative mechanism at early stage, but at late stage, diabetes not only generates extra ROS and/or RNS but also impairs antioxidant capacity in the heart [4,9]. Decreased expression of heat shock protein 60 and heme oxygenase-1 in the diabetic heart made the heart high susceptible to oxidative damage [9,10,11]. Therefore, antioxidant therapy for DCM has been attractive, but its outcomes to prevent cardiac complications in diabetic patients by dietary supplementation of antioxidants are controversial [12,13,14]. Several reasons including the difficulty in maintaining a consistent circulating antioxidant levels for supplied exogenous antioxidants, inadequate tissue distribution, and lack of suitable exogenous antioxidants have been discussed [3,14,15]. This might be one of the reasons that single or a few of antioxidants together would remove limited kinds of free radicals while diabetes can induce multiple kinds of free radicals.
This concept may be implicated by the action of metallothionein (MT). MT is a cysteine-rich (1/3 of 61 amino acids) and binds zinc under physiological condition [16,17,18]. As a nonspecific antioxidant, MT is able to quench a wide of free radicals, including superoxide, hydrogen peroxide, hydroxyl radical, and peroxynitrite [16,17,18]. We have shown that MT as a potent, nonspecific antioxidant significantly prevented various diabetic complications in animal models and human, suggesting its great potential for clinical application to prevent DCM [5,15,19,20,21,22,23,24]. However, MT remains a single antioxidant, which promotes us to further look for an approach to up-regulating multiple antioxidants, including MT, for efficiently preventing diabetic complications.
THE TRANSCRIPTION FACTOR NUCLEAR Nrf2
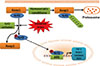
Cells contain a number of genes coding many proteins to counteract ROS-, RNS-, or electrophile-mediated injury. Transcriptional regulation of these protective genes is controlled in part through antioxidant response elements (AREs) [25,26]. The transcription factor nuclear factor NF-E2-related factor 2 (Nrf2) plays an important role in ARE-mediated basal and inducible expression of more than 200 genes that can be grouped into several categories including antioxidant genes and phase II detoxifying enzymes [25,26], as outlined in Fig. 2. These antioxidant components include heme oxygenase-1, thioredoxin reductase, glutathione-S-transferase, and NAD(P)H:quinone oxidoreductase (NQO)-1, antioxidant enzymes such as superoxide dismutase and catalase, and nonenzymatic scavengers such as glutathione. The protein stability and transcriptional activity of Nrf2 is principally regulated by a BTB-Kelch protein, Keap1 that functions as a substrate adaptor for a cullin (Cul)3-dependent E3 ubiquitin ligase complex. Keap1 targets Nrf2 for ubiquitination and subsequent degradation by the 26S proteasome [25,26].
In an early study, we have indicated the important role of Nrf2 in preventing high glucose-induced production of ROS and apoptotic cell death in both primary neonatal and adult cardiomyocytes from the mice with deletion of the Nrf2 gene (Nrf2-KO) than those from the Nrf2 wild-type mice [27]. Primary adult cardiomyocytes from Nrf2-KO diabetic mice showed a loss of isoproterenol-stimulated contraction compared to those from Nrf2 wild-type diabetic mice. Our finding was the first one to establish Nrf2 as a critical regulator of defense against ROS in normal and diabetic hearts [27]. We further showed the significant down-regulation of cardiac Nrf2 expression in diabetic animals and patients [28]. Now it is clear that down-regulation of Nrf2 is a significant reason for the initiation of various diabetic complications [29,30]. Using Nrf2-KO mice or Nrf2 inducers blooming evidence has indicated the protection by Nrf2 from diabetes [31,32] and from various diabetic complications [33,34,35,36,37]. We have worked on the prevention of DCM and other diabetic complications with several Nrf2 inducers [33,35,38,39,40,41,42], as summarized in Table 1 [32,35,36,37,39,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60].
The effective protection from diabetic complications by up-regulating Nrf2 function in animal models promoted clinical trials with Nrf2 inducer to prevent diabetic nephropathy. In 2011, the phase II clinical trial by following-up 52 weeks for the treatment of participants with moderate-to-severe diabetic kidney disease with bardoxolone methyl (BM) [61] reported the improvement of renal function compared to non-BM-treated diabetic patients. But the phase 3 clinical trial for the patients with advanced diabetic kidney disease [62] was prematurely terminated due to the strong adverse effects associated with BM treatment, including increased rates of heart failure and cardiovascular events. The failure of BM clinical trial suggests that more detail study in preclinical animal models is urgently needed before new clinical trials. Given the efficient prevention of diabetic complications with Nrf2 inducers in various animal models and the escalating human and societal costs of diabetic complications, efforts to find new safe and effective drugs via up-regulating Nrf2 remain vital [63,64,65,66].
Monascus-fermented metabolite monascin acts as a novel natural peroxisome proliferator-activated receptor-γ (PPARγ) agonist that improves insulin sensitivity, but dislike rosiglitazone, monascin was also able to activate Nrf2 to further elevate glyoxalase-1 expression. Monascin may be a novel natural Nrf2 activator with PPARγ-agonist activity. Therefore, monascin acts as an antidiabetic and antioxidative stress agent to a greater degree than rosiglitazone and thus may have therapeutic potential for the prevention of diabetes [67].
UNDESIRABLE SIDE OF Nrf2
It should be mentioned that everything has both sides; Nrf2 also has its undesirable side. It is well-known that ROS derived from multiple sources plays a causal role in multiple types of insulin resistance and contributes to β-cell dysfunction, leading to enhance the development and progression of type 2 diabetes, in another word: the detrimental ROS also plays a substantial role in the normal insulin signal transduction and glucose-stimulated insulin secretion in β-cell. Therefore persistent activation of Nrf2 gene may cause over-reduction of these required ROS signaling in the body. Consequently the detrimental effects of Nrf2 due to its aberrant activation have also been highlighted in recent years. A few of such examples include: (1) constitutive Nrf2 activation worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin-deficient mice [68]; (2) Nrf2 deficiency improves glucose tolerance in mice fed a high-fat diet [69]; and (3) Nrf2 deficiency prevents reductive stress-induced hypertrophic cardiomyopathy [70,71]. For instance, the latter cases highlighted that certain amount intracellular oxidative modification of proteins is a key event required for proper ubiquitination and protein degradation [72]. If substantial activation of Nrf2 may cause a significant decrease of protein oxidation in association with the induction of chronic reducing stress, which in turn causes deubiquitination and downstream protein degradation pathways, resulting in the development of cardiac hypertrophy and remodeling [70,71].
In addition, although the undesirable side of Nrf2 has been extensively mentioned in the cancer research field, Nrf2 has been shown to protect normal cells from tumor formation by decreasing the oxidative stress and preventing the DNA damage in cells. However, recently the cancer-promoting role of Nrf2 has been revealed. Nrf2 was found to be constitutively up-regulated in several types of human cancer tissues and cancer cell lines, and to protect tumors and cell lines from chemotherapeutic drugs [73,74].
CONCLUSIONS
In summary, the development and progression of DCM, one of the major cardiac complications in diabetic patients, is predominantly related to oxidative stress that is due to a severe imbalance between ROS and/or RNS generation and their clearance by antioxidant defense systems. In our body, cells have a well-established defense system against oxidative stress, such as Nrf2 that plays an important role in maintaining the oxidative homeostasis by regulating multiple downstream antioxidants. Diabetes, at late stage, not only generates extra ROS and/or RNS but also impairs antioxidant capacity in the heart, including Nrf2. We have demonstrated that Nrf2 protects the cardiac cells and heart from high level of glucose in vitro and hyperglycemia in vivo, respectively. We also found that diabetes significantly down-regulated cardiac Nrf2 expression in diabetic animals and patients, which might explain the development and progression of DCM. By using Nrf2-KO mouse model, the important role of Nrf2 in protecting various organs including the heart from diabetes has been extensively approved. Therefore various Nrf2 inducers have been explored with the objective of developing an applicable approach to pharmacologically up-regulate our systemic levels of Nrf2 in diabetic individuals for a prevention of their complications. Although the first candidate, bardoxolone methyl, has been failed in the phase II clinical trial that showed the potent cardiac toxicity when bardoxolone methyl was applied to diabetic patients with renal dysfunction, this could not stop searching for an efficient and safe new Nrf2 inducer. In addition, it also gradually is realized that anything can not be too much even though it is beneficial to us at most time. Similarly Nrf2 was found to also have its undesirable side when it is substantially and overexpressed. Therefore, when we continually explore for the approach to enhancing organ's Nrf2 expression with the purpose of prevention of cardiovascular complications, we also should keep in mind of its undesirable side under certain conditions.

 XML Download
XML Download