

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Extensive epidemiological studies suggest that certain types of cancers show a higher prevalence rate and a higher risk of mortality in a patient population suffering from diabetes mellitus (DM) [1]. Liver and pancreatic cancers show a strong relationship with DM, and both of these organs play a central role in the pathophysiology of diabetes [1,2,3]. Other cancers, such as colorectal, breast, endometrial and renal, and renal cancer also show an association with DM, whereas prostate cancer appears to have a negative association with DM [4]. Thus, it is now evident that a higher cancer risk and mortality rate is observed in DM patients compared to euglycemic individuals. Furthermore, DM and cancer are frequently diagnosed in the same individuals [5], suggesting that those two diseases share the common risk factors and pathophysiological mechanisms. The underlying mechanisms behind this association have not been fully elucidated; however, plausible connections consist of hyperinsulinemia, insulin resistance, chronic inflammation, oxidative stress, and hyperglycemia; all of these factors potentially promote tumor progression in various ways [6,7,8,9,10]. The impact of insulin, insulin-like growth factor-1 (IGF-1) and chronic inflammation in cancer progression has been extensively studied, whereas effects of hyperglycemia on cancer have received less attention, although hyperglycemia is one of the most widely studied metabolic changes in DM.
Hyperglycemia is defined as a state of excess glucose concentration in circulation, a hallmark for both type 1 DM and type 2 DM. Due to insufficient insulin production in pancreatic β cells, hyperglycemia develops in type 1 DM. Additionally, the increase of systemic insulin resistance in type 2 DM leads to hyperglycemia as well [1]. Hyperglycemia indirectly influences cancer cells through an increase in the levels of insulin/IGF-1 and inflammatory cytokines in circulation. Beyond that, there are reasons to believe that hyperglycemia per se has a direct impact on cancer cell proliferation, apoptosis and metastasis [1,2,3]. High glucose activates various signaling pathways that cooperate to control cancer cell behavior, such as proliferation, migration, invasion and recurrence [11]. Furthermore, epigenetic modulations of oncogenic pathways induced by high glucose result in prolonged activation of cancer cell proliferation [12,13]. However, these direct effects of high glucose in cancer cell behavior are relatively unexplored, and more needs to be learned about which signaling pathways are involved and how they are controlled. Resolving these questions will shed light on the molecular links between DM and cancers. Below, we will discuss the potential mechanisms linking hyperglycemia to cancer progression, with a special focus on tumor cell proliferation, apoptosis, invasion and the epigenetic variations in cancer cells upon hyperglycemic exposure. This will by necessity also require a discussion of the efficacy of DM therapeutics for cancer growth.
IMPACT OF HYPERGLYCEMIA ON CANCER CELL BEHAVIOR
Cancer cell proliferation
Enhanced glucose uptake in cancer cells is a well-established hallmark of cancer cells [14]. The enhanced glucose metabolism in cancer cells is referred to as the Warburg effect, which comprises the increase in aerobic glycolysis in cancer cells defined by Warburg [15]. In this respect, hyperglycemia could provide a high glucose fuel source for cancer cells supporting rapid proliferation. Indeed, in vitro studies with cancer cell lines indicate that high concentrations of glucose levels regulate enhanced expression of genes associated with promoting cancer cell proliferation, invasion, and migration [16]. In vivo animal studies are plagued by technical limitations to evaluate the effects of hyperglycemia on cancer cells. This is due to the fact that in order to induce hyperglycemia in these rodent models, streptozotocin is used to destroy pancreatic β-cells. This leads to hyperglycemia, but obviously also prompts the loss of insulin in circulation. Insulin is an intrinsic mitogen affecting tumor growth. In humans, elevated glucose levels in circulation serve as an established predictor of poor survival in cancer patients [17,18,19]. In line with that, high sugar intake is associated with increased cancer cell proliferation [16].
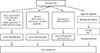
Multiple proteins have been implicated as mediators of hyperglycemia and cancer cell proliferation (Fig. 1). Recent in vitro studies suggest that the expression of glucose transporters, such as the GLUT1 and GLUT3 isoforms, is regulated under hyperglycemic conditions in JAR cells, a choriocarcinoma cell line [20]. Hyperglycemic conditions in vitro (such as 25 mM D-glucose) trigger increased glucose uptake in JAR cells due to a transcriptional increase and enhanced protein levels for GLUT1 and GLUT3 [20]. Growth factors, such as epidermal growth factor (EGF) levels are augmented by high glucose treatment in pancreatic cancer cell lines, such as BxPC-3 and Panc-1, and subsequently activate its receptor, the epidermal growth factor receptor (EGFR), a well-known oncogenic pathway [21]. In addition, the levels of protein kinase C (PKC) and peroxisome proliferator-activated receptors (PPARs) are stimulated under hyperglycemic conditions in MCF-7 human breast cancer cells [22]. Overexpression of PKC-α in MCF-7 induces a more aggressive phenotype [23]. PPAR-α and PPAR-γ are influencing lipid metabolic pathways. High levels of PPAR-α and PPAR-γ can accelerate cell proliferation [24]. High glucose accelerates the cell cycle through regulating the levels of key proteins, such as cyclin-dependent kinase 2, E2F, cyclinA, and cyclinE, resulting in increased proliferation [16]. Furthermore, hyperglycemic conditions augment the levels of glial cell line derived neurotophic factor (GDNF) and its tyrosine kinase receptor gene, the rearranged during transfection (RET) gene, in human pancreatic cancer cells, such as BxPC-3 and MIA PaCa-2 cells [25]. GDNF is a cytokine related to the transforming growth factor-β family, enhancing the survival and differentiation of midbrain dopaminergic neurons [26] as well as promoting pancreatic cancer cell proliferation and invasion mediated through a protein complex of RET/GDNF-family receptor α-1 (GFR α-1) [27]. Taken together, these data strongly implicate hyperglycemia as a contributing factor leading to enhanced cell proliferation.
Cancer cell antiapoptosis
Apoptosis, the process of programmed cell death, is a genetically regulated process that is essential for multicellular organisms. The dysregulation of apoptosis can result in uncontrolled cellular growth [28]. The connection between hyperglycemia and cancer cell apoptosis is unclear; however, there are several possible connections suggested (Fig. 1).
Many tumors are exposed to hypoxia due to limited oxygen supplies during rapid anabolic cell proliferation [29]. In response to hypoxia, hypoxia inducible factor-1α (HIF1α), a key transcriptional regulator of the hypoxic response, is stabilized at the protein level and translocated into nucleus. This leads to the increased expression of genes associated with glucose metabolism, angiogenesis and survival/antiapoptotic processes [30,31]. Under normoxic conditions, HIF1α is degraded by HIF prolyl hydroxylase (PHD) enzymes, and this process is oxygen dependent [31]. Hyperglycemia regulates the stability and function of HIF1α through interfering with the degradation of HIF1α by PHD enzymes [32], which causes increased cancer cell survival and conveys antiapoptotic qualities upon the tumor.
Recent studies also suggest that glucose metabolism in cancer cells protects cytochrome c-mediated apoptosis [33]. Glutathione is one of the main antioxidant mediators of cells, which is reduced by nicotinamide adenine dinucleotide phosphate (NADPH) derived from enhanced glucose metabolism, such as the pentose phosphate pathway [34].
Cancer cell migration and invasiveness
Epidemiological data suggest that metastatic growth is the main cause of deaths in 90% patients carrying solid tumors [35]. Metastatic growth ensues when cancer cells become invasive through an altered phenotype, penetrating into the circulatory system, and taking hold in a distant organ. Epithelial-mesenchymal transition (EMT), a multifaceted process critical for the acquisition of migration, invasiveness and pluripotent stem cell-like phenotype, plays a pivotal role in the metastatic process [36]. Several studies suggest that high glucose induces cancer cell invasiveness and migration through stimulating EMT (Fig. 1). High glucose decreases E-cadherin levels, an epithelial cell marker, and increases the PKC-α pathway, leading to a more invasive phenotype [16]. Recently, Dong et al. [37] suggested that hyperglycemia induces the EMT phenotype and the expression of cancer stem cell markers in basal luminal breast carcinoma, which leads to reduced reactive oxygen species (ROS) generation and increased cell survival.
Hydrogen peroxide has been implicated in the migration and invasive activity of pancreatic cancer cells under hyperglycemic conditions [38]. High glucose levels generate oxidative stress due to ROS. The migration and invasion of pancreatic cancer cell lines, such as BxPC-3 and Panc-1 cells, are all augmented by superoxide dismutase (SOD) which catalyzes the conversion of the superoxide anion to hydrogen peroxide under hyperglycemic conditions [39]. The mRNA expression of urokinase plasminogen activator, one of the mediators involved in cell migration, is also up-regulated under high glucose levels with SOD [40].
Zinc is an essential element for cellular function. An imbalance in zinc homeostasis causes several human diseases [41]. Hyperglycemia is thought to promote migration of breast cancer cells via zinc and its transporters, ZRT/IRT-like protein 6 (ZIP6) and ZRT/IRT-like protein 10 (ZIP10) [42]. Thus, high glucose increases zinc uptake, and the expression of ZIP6 and ZIP10 transporters in breast cancer cells, such as MCF-7. ZIP6 regulates EMT, and ZIP10 is known to be involved in cancer cell migration [43].
Epigenetic regulation in cancer cells
Chronic hyperglycemic conditions may cause epigenetic changes in oncogenic pathways in cancer cells (Fig. 1). A recent study suggested that epigenetic silencing of the critical gluconeogenic enzyme, fructose-1,6-biphosphatase through the EMT-related transcription repressor Snail, increases glycolysis and NADPH production via the pentose phosphate pathway and a reduction in oxidative phosphorylation [37]. These metabolic alterations induced by hyperglycemia contribute to lower ROS generation and increase of β-catenin/TCF4 activation, a key pathway for the acquisition of a cancer stem cell phenotype, leading to better survival [37]. Recent studies show that transient hyperglycemia induces the recruitment of the transcription factor Set7 to the nuclear factor-κB (NF-κB) p65 promoter. The recruitment of Set7 to p65 promoter enhances histone 3 lysine 4 monomethylation within the promoter, resulting in increased NF-κB activation and increased inflammation [44].
Hyperglycemic memory is an epigenetic phenomenon induced by hyperglycemia [12]. After cancer cells are exposed to hyperglycemic conditions, a subset of oncogenic pathways are permanently activated, even after hyperglycemic conditions are normalized to euglycemic conditions [45]. However, the specific molecular mechanism how a cancer cell gets permanently "rewired" and the hyperglycemic memory instilled is still unclear. We suggested that the neuregulin-1 (Nrg1)-HER3 pathway is up-regulated in tumors derived from hyperglycemic patients or rodents. Nrg1 belongs to a family of EGF-like ligands for HER3, itself a member of the EGFR of receptor tyrosine kinase, related to cancer cell proliferation, survival and metastasis [46]. Once activated, these cancer cells originating from hyperglycemic conditions grow faster than control cells even under euglycemic conditions [45].
Therapeutic interventions
Given these strong epidemiological connections seen in patients suffering from DM with a higher risk to develop some types of cancer, these increased risks need to be taken into account and prophylactic screening is warranted. However, an additional question is whether antidiabetic therapeutic approaches also reduce the risk of associated cancers.
There are a variety of antidiabetic interventions, including sulfonylureas, α-glucosidase inhibitors, biguanides, and thiazolidinediones (TZDs) [47]. The major groups of antidiabetic drugs increase the level of circulating insulin, thereby reducing hyperglycemia by various mechanisms. Especially, compounds such as metformin inhibit hepatic gluconeogenesis to reduce the level of circulating glucose and increasing insulin sensitivity through the 5'AMP-activated protein kinase and AKT/mTOR pathway [48,49,50]. Metformin has received widespread attention due to its apparent anticancer effects [48,51]. Metformin inhibits cell proliferation and induces apoptosis in cancer cell lines [52,53,54]. It can also exert anticancer effects via modulation of DICER (also known as endoribonuclease Dicer or helicase with RNase motif) activity, via mir33a up-regulation, and via targeting c-MYC (Myc proto-oncogene protein) [55]. Despite a lot of excitement and attention, clinical data inferring anti-mitogenic effects of antidiabetic drugs and their effects on cancer incidence and mortality remain somewhat controversial. As such, some studies suggest that insulin and insulin glargine can increase cancer incidence [56,57], while other studies conclude that there is no effect [58]. Similarly, TZDs have been reported to exert both tumor growth supporting and inhibitory roles [59], whereas metformin and other biguanides decrease the cancer incidence [60,61] or have no impact [62].
Another drawback of treating cancers in the context of DM is at the level of the memory effects of hyperglycemia and the associated epigenetic alterations in key oncogenic pathways in cancer cells. Thus, merely managing blood glucose levels per se might not be sufficient to obtain a full therapeutic impact. We need to expand our understanding of the key specific target genes and pathways affected by hyperglycemia within tumors, such that these pathways may be targeted in a more directed approach. For instance, the finding that the NRG1/HER3 axis is specifically and disproportionately activated in women diagnosed with breast cancer and found to be hyperglycemic at the time of diagnosis may suggest an effective first line of treatment with neutralizing Her3 antibodies which would not necessarily be the first choice in the context of other types of breast cancer.
CONCLUSIONS
Strong epidemiological data is at hand suggesting an increase of both cancer risk and mortality in DM patients. This is especially relevant for cancers of the liver, pancreas, mammary gland, and the endometrium. Studies have mainly focused on the molecular mechanisms related to insulin and chronic inflammation. Hyperglycemia and associated hyperglycemic memory effects and their impact on key cancer cell pathways, such as cell proliferation, apoptosis, migration, and invasion offer new therapeutic avenues. These combined with traditional chemotherapeutic approaches may offer strong synergistic effects towards curbing growth of primary tumors and metastatic lesions. Diabetic patients may profit from these more individualized treatment regimens. Unquestionably, with the ever increasing prevalence of diabetes in the population, affecting younger individuals more than ever before, the interface of metabolic dysregulation and cancer moves increasingly to the forefront and represents as much of a challenge as it is an opportunity for novel therapeutic approaches.
 XML Download
XML Download