

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Under normal conditions, neurons, glia, and vasculature of the brain or retina act together in a coordinated manner. Cells constantly are bathed in chemical moderators, hormones, ions, and proteins from surrounding cells and tissues, and these signals coordinate a local interrelationship between metabolism, capillary density, and blood flow. Notably, changes in neuronal activity induce changes in vascular blood flow and density.
The purpose of this review is to summarize data indicating that the interaction between neural and vascular cells becomes altered in retina in diabetes, and to provide new evidence that suggests that neural cells of the retina can contribute to the development of vascular alterations that are characteristic of the early stages of diabetic retinopathy.
NEUROVASCULAR COUPLING
Neurovascular coupling refers to the relationship between local neural activity and changes in blood flow. Thus, this describes a process in which neural tissue regulates its blood flow in response to neural activity.
In the retina, there is significant linkage between neural activity of retinal ganglion cells (or at least inner retina) and blood flow [1,2,3,4]. The retina has unique sensitivity to light, and the response of the retinal vasculature to light-induced changes in neural function has provided considerable insight into neurovascular coupling in the retina [4]. Human and animal studies have indicated that retinal or photoreceptor oxygen consumption is greater in dark than light, yet oxygen saturation in retinal blood vessels is higher in dark than in light [5]. Moreover, velocity of blood in the retinal vessels is increased at night [6]. The increased delivery of oxygen in response to the increased activity of retinal neurons is a consequence of the coupling between neural activity and the vasculature. Since the blood vessels do not directly respond to light, light-induced changes in blood flow or oxygen delivery must be initiated by light-induced changes in neural activity of the retina. Surprisingly, dark adaptation did not increase retinal blood flow in Sprague-Dawley rats.
Flicker light stimulation further demonstrates neurovascular coupling in the retina. Various animal and human studies have demonstrated that retinal and optic nerve blood flow increase in response to diffuse luminance flicker (light on, light off) [7]. Diffuse luminance flicker stimulation increased optic nerve-head blood flow markedly in anesthetized cats, and the increase depended upon the intensity, frequency, and wavelength of the stimulation and the state of adaptation of the retina [8]. Blood flow around the macula, as assessed by blue field simulation technique in normal subjects, also increases in response to diffuse luminance flicker [9]. Investigators have reported that flicker light-induced vasodilation was mediated by ganglion cells [4] or pericytes [10]. There is significant correlation between neural activity of retinal ganglion cells and enhanced glucose delivery and metabolism during light flicker [11].
Neurovascular communication
Possible mechanisms by which neuroglial cells communicate with vascular cells have been investigated. Some of these signaling molecules act rapidly (such as H+, K+, neurotransmitters, adenosine, arachidonic acid metabolites, and nitric oxide [NO]), whereas others seem to have a more prolonged effect (such as growth factors) [4,12,13]. It seems clear that the control of microcirculation by neuroglia is a complex process that likely involves a number of different molecules [14]. Some of these molecules are listed here.
1) NO can be made by every retinal cell type, and it is an important signaling molecule that regulates neurotransmitters release and modulates gap junction conductivity in retina. In the retina, the NO produced by the constitutive nitric oxide synthases (NOSs; endothelial NOS and neuronal NOS) contribute to regulate normal ocular hemodynamics and cell viability, and to protect retinal cells against different stresses [15,16]. Pretreatment of animals with a NO inhibitor inhibited the flicker-induced increase in retinal blood flow [17], indicating that NO plays an important role in the coupling between retinal neuronal activity and blood flow.
2) Metea and Newman [18] demonstrated that retinal glial cells mediate neurovascular coupling by inducing the production of two arachidonic acid metabolites, epoxyeicosatrienoic acids, and 20-hydroxyeicosatetraenoic acid, which dilate and constrict vessels, respectively. They showed that 1) light or direct glial stimulation produce vasodilatation or vasoconstriction mediated by these small molecules, 2) the vascular response depends on NO, and 3) glial cells are necessary for signaling from neurons to blood vessels.
3) Studies have demonstrated that a large fraction of neurovascular coupling that causes hyperemia is due to release of glutamate from neural tissue [19].
4) Vascular endothelial growth factor (VEGF) is another soluble factor that couples neuroglia and the vasculature. VEGF is produced largely by glia and neurons. It is known to have important effects on vascular development, permeability and, in ischemic diseases, also neovascularization [20].
5) Neurotrophins are another way by which neural tissue might communicate with the retinal vasculature. The neurotrophins are a family of trophic factors (including nerve growth factor [NGF], brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4/5) which promote neural cell survival, neurite outgrowth, phenotypic maturation, and synaptic functioning, but also exert a trophic effect on some, but perhaps not all, endothelial cells. Endothelial cells from the brain, dermis, and umbilical vein have been reported to contain the NGF receptors TrkA and p75NGFR, and to respond to NGF with cell mitosis, migration, and survival. Cerebral endothelial cells, however, have been reported to not respond to NGF [21].
NEUROVASCULAR INTERACTIONS AND DISEASE
Alterations in neurovascular coupling and cerebrovascular regulation have been proposed to play a key role in the process of ageing as well as in stroke, hyperlipidemia, hypertension, and diabetes. This review will now focus on alterations in neurovascular interactions in diabetes.
Regulation of retinal blood flow by neural activity in diabetes
Whereas a variety of studies describe the effects of flickering light on retinal and optic nerve head blood flow under normal conditions, the knowledge about this coupling in the diabetic retina is less complete. Nevertheless, available evidence indicates that neurovascular coupling in the retina is altered in diabetes. Luu et al. [22] demonstrated that a component of the neural oscillatory potential of the electroretinogram (ERG) correlated with retinal arteriolar caliber in patients with diabetes, demonstrating at least a correlation between retinal neuronal dysfunction and blood flow. Flicker responses of retinal arteries and veins in insulin-dependent diabetic patients [23] and in healthy subjects made experimentally hyperglycemic [24] have shown significant reduction compared to controls. The flicker-induced retinal diameter change has been shown to deteriorate early in patients with diabetes; Lecleire-Collet et al. [25] found a significant reduction of the flicker-induced response in the retinal arteries and veins of normotensive patients with diabetes with no clinically detectable diabetic retinopathy. Nguyen et al. [26] showed that reduced retinal vasodilation after flicker light stimulation was associated with diabetes, independent of major risk factors such as hypertension and glycemic control. Whether or not there is a difference between type 1 diabetes and type 2 is not clear; a controlled study demonstrated decreases in both arterial and venous flicker-induced retinal vasodilation in normotensive patients with type 1 diabetes and no diabetic retinopathy [27], whereas an uncontrolled study of patients with type 2 diabetes by the same research group [28] demonstrated that the observed decreases in the arterial and venous flicker-induced responses were no longer significant after factoring out age and antihypertensive treatment.
The impaired flicker-induced vascular response may be partly caused by endothelial dysfunction, as suggested by Mandecka et al. [27,28] and Nguyen et al. [26,29]. A key role for NO has been identified in retinal and optic nerve vasodilatory response to greater neuronal activity. Light-evoked arteriole dilation (functional hyperemia) is reduced diabetic rats, and inhibition of iNOS restored both light- and glial-evoked dilation of retinal arterioles to control levels [30].
Diabetic retinopathy
Diabetic retinopathy is a common complication of diabetes and a leading cause of legal blindness among working-age adults. Historically, diabetic retinopathy researchers and physicians have concentrated on the vascular lesions in this disease, since those are what seem to account for most of the lesions that contribute to vision loss in diabetes. It is now clear that the neuronal components of the retina are also injured in diabetes, and appear to become impaired prior to the regression of the retinal vasculature. We will first present a brief summary of diabetic retinopathy, and then summarize recent evidence that neural abnormalities within the retina are actually contributing to the vascular lesions of the retinopathy.
Vasculature
Diabetic retinopathy is comprised of a spectrum of histologic and functional abnormalities that in composite make a picture that is relatively unique for diabetes. These abnormalities include nonperfused and degenerate capillaries, pericyte ghosts (empty pockets in the basement membrane surrounding capillaries where pericytes used to be located), microaneurysms, increased vascular permeability, and intraretinal microvascular abnormalities in early stages of the disease. In the advanced (clinically important) stages of the retinopathy, hemorrhage, retinal edema, neovascularization, and possibly retinal detachment occur.
Neurons
Some nonvascular cells, likely retinal ganglion cells, begin to degenerate (become positive for terminal deoxynucleotidyl transferase dUTP nick-end labeling stain (TUNEL) early in diabetes, and in fact some retinal neurons apparently begin to degenerate earlier than do vascular cells [31,32,33]. Retinal cross-sections or in vivo measurements from diabetic rodents or patients have revealed retinal thinning in diabetes [33], which also is consistent with neuronal death. Although some neuroglial cells do die in diabetes, even greater numbers of cells likely survive but become dysfunctional.
Diabetes-induced retinal dysfunction has been well documented. In the retinas of human diabetics, pattern ERGs become diminished before the onset of the classically defined nonproliferative retinopathy [34,35], and delayed physiological signals (evoked potentials) have been found in the optic nerve (axons of the retinal ganglion cells) and visual pathways of diabetic patients. A recent study of early diabetes indicated that patients having nonproliferative diabetic retinopathy showed impairment of visual function compared to normal participants, with 83% of patients exhibiting clinically significant impairment [36]. In addition, rod photoreceptor function was grossly impaired. Contrast sensitivity and other defects in vision have been widely studied in patients with diabetic retinopathy. Trick et al. [37] found that about one-third of diabetic subjects without retinopathy had abnormalities in contrast sensitivity, this figure rising to about two-thirds in those with nonproliferative (background) retinopathy. These electrophysiological measures provide indications of functional changes in neural cells of the retina in the earliest stages of human diabetic retinopathy.
Retinal levels of NO, glutamate, VEGF and neurotrophins are altered in diabetic retinopathy.
1) Nitric oxide
It is clear from multiple investigations that NO levels are altered in diabetes. Increased serum levels of nitrite and nitrate (breakdown products of NO) have been found in diabetic patients with retinopathy compared with diabetic patients without diabetic retinopathy or healthy controls. In those studies, serum nitrite and nitrate (used to estimate NO) levels in the patients with proliferative diabetic retinopathy were significantly higher than the levels in the patients with nonproliferative retinopathy. Likewise, plasma nitrite and nitrate levels were higher in patients with proliferative diabetic retinopathy than in controls also in other studies. Elevated metabolites of the L-arginine-NO pathway have been detected also in the vitreous of eyes from diabetic patients, and greater than normal levels of NG-hydroxy-L-arginine (a by-product of NO generation), have been detected in the ocular aqueous humor of diabetic patients (with and without diabetic retinopathy) compared with that in nondiabetic controls. Despite what appears to be increased generation of NO in diabetes; however, diabetes results in subnormal NO bioavailability, due to increased production of free radicals which directly react with NO to generated peroxynitrite, or oxidize the cofactors of the NO synthase, diminishing the activity of NO synthases and consequently leading to a decreased NO production [38].
2) Glutamate
In diabetes, levels of the excitotoxin, glutamate, have been reported to be significantly increased [39] or decreased [40] in retinas of diabetic rodents. In diabetic patients, levels of glutamate in the vitreous are significantly elevated [41,42]. The increase in retinal glutamate in diabetic rodents was inhibited with systemic administration of antioxidants [39].
3) VEGF
Levels of VEGF in retina and vitreous are elevated in diabetes, and there is strong evidence that this VEGF increases vascular permeability and neovascularization in advanced diabetic retinopathy [43,44]. In vitro, VEGF plays a critical role in endothelial cell apoptosis induced by high glucose [45]. In vivo, Bai et al. [46] demonstrated that disruption of Müller cell-derived VEGF resulted in significant inhibition of the ischemia-induced retinal neovascularization and vascular leakage, and attenuation of the ischemia-induced breakdown of the blood-retina barrier.
4) Neurotrophins
Systemic administration of NGF to diabetic animals inhibits diabetes-induced vascular degeneration in the retina [47]. This benefit of NGF administration might not be mediated by a direct effect on the endothelium itself, but rather an indirect effect on other members of the neurovascular components in the retina, because retinal endothelial cells have been reported not respond to NGF in vitro [48]. The observed beneficial effect of NGF on the retinal vasculature in diabetes might indicate a diabetes-induced defect in neurovascular communication that can be corrected therapeutically. Diabetes-induced impairment of NGF processing leads to accumulation of pro-NGF, which has been postulated to contribute to development of diabetic retinopathy [49].
NEW EVIDENCE SUGGESTING THAT RETINAL NEURONS CONTRIBUTE TO VASCULAR DISEASE IN DIABETIC RETINOPATHY
The finding that retinal neurons (especially ganglion cells) began dying (estimated from TUNEL staining) before vascular lesions were grossly apparent led some investigators to speculate that neurons might contribute to the development of the vascular lesions of diabetic retinopathy [31]. No evidence to support this postulate was identified until recently, when retinal photoreceptors were identified as likely contributors to the vascular damage in the retinopathy.
Photoreceptors are specialized retinal neurons, and mediate the capture of light energy and conversion of that light into neural signals that allow us to see. The high metabolic rate of photoreceptors has been recognized as contributing to development of retinal hypoxia and subsequently neovascularization in late, advanced stages of diabetic retinopathy, but recent reports have raised a possibility that photoreceptors or outer retina might play a role in the development also of even early stages of diabetic retinopathy. de Gooyer and collaborators [50] reported that diabetes did not cause the expected decrease in density of the retinal microvasculature in mice lacking rhodopsin (Rho-/-, which causes photoreceptor degeneration). The authors concluded that loss of the outer retina reduced the severity of diabetic retinopathy in that model. Two additional pieces of data involving diabetic patients are consistent with a role of photoreceptors in the development of diabetic retinopathy. Results of a survey sent to a small group of diabetic patients who also had retinitis pigmentosa (and therefore, photoreceptor degeneration) suggested that diabetic retinopathy was less severe in patients who also had retinitis pigmentosa [51]. In addition, Arden et al. [52,53] conducted a study on patients to determine if sleeping without total darkness (to reduce the dark current in rod photoreceptors) could improve diabetes-induced abnormalities of retinal function or structure, and found that the avoidance of total darkness significantly reduced the tritan thresholds, the area of dark retinal anomalies, and retinal edema in treated eyes relative to untreated eyes.
The mechanism by which neurons might contribute to vascular pathology of diabetic retinopathy will be important to determine. Perhaps impaired delivery of oxygen to photoreceptors at night and the resulting low oxygen levels in the retina, lead to increased levels of VEGF and its effects on the vasculature. In addition, recent studies demonstrate that photoreceptors generate (or at least regulate) generation of reactive oxygen species and synthesis of inflammatory proteins in the retina in diabetes [54]. This is important because diabetes results in increased generation of superoxide and inflammatory proteins in retina, and inhibition of the oxidative stress or deletion/inhibition of inflammatory proteins has been shown to inhibit development of vascular lesions of early diabetic retinopathy [55,56]. Low intensity light is being found to inhibit aspects of diabetic retinopathy [52,53,57], and offers a possible therapeutic direction for future efforts to inhibit the retinopathy.
CONCLUSIONS
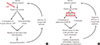
Clinical evidence continues to support the premise that retinal vascular disease accounts for much of the clinically significant loss of vision that occurs in diabetes. However, neural function is an important regulator of normal vascular function, and disturbances in the interaction between neural and vascular cells can have adverse effects on retinal function or structure (Fig. 1). Intriguing new evidence indicates that photoreceptors and other neural cells might contribute to the pathogenesis of the vascular disease that is characteristic of diabetic retinopathy. Thus, therapeutic efforts to correct critical abnormalities within the retinal neuroglia might help inhibit both the vascular lesions that characterize diabetic retinopathy and the diabetes-induced loss of vision. Additional work is required to further investigate this possibility.
 XML Download
XML Download