

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The incidence and prevalence of diabetes mellitus have significantly increased worldwide in recent decades, primarily due to the increase in type 2 diabetes mellitus (T2DM). Long-term diabetes results in vascular changes and dysfunction, and diabetic vascular complications are the major cause of morbidity and mortality in patients with diabetes. It is not a simple matter to strictly control blood glucose levels for long periods of time, even given the many antidiabetic medications that are clinically available. Therefore, the development of improved additional treatments and novel prevention strategies for T2DM is a matter of great urgency.
Aging is implicated in metabolic diseases, including diabetes; therefore, aging is recognized as a risk factor for the initiation and the development of T2DM. Calorie restriction (CR) without malnutrition promotes longevity and slows aging. Numerous studies have revealed that CR retards aging or extends the lifespans of yeast, worms, flies, and rodents. Colman et al. [1] also reported that 30% CR delayed the onset of numerous age-associated pathologies, including diabetes, cancer, cardiovascular disease and brain atrophy, and decreased mortality in rhesus monkeys. Moreover, Fontana et al. [2] reported that CR for an average of 6 years improved metabolism in humans, as was indicated by levels of serum insulin, cholesterol, C-reactive protein (CRP) and tumor necrosis factor (TNF)-α as well as by carotid intima media thickness. This group also observed that long-term CR ameliorated the decline in left ventricular diastolic function and decreased levels of serum tumor growth factor-β1, TNF-α, and high-sensitivity CRP [3]. Thus, CR has a variety of beneficial effects with respect to lifespan extension and delays the onset of age-related diseases, such as cardiovascular diseases, neurodegenerative disorders, and diabetes. CR is defined as the restriction of food intake without malnutrition in organisms that are normally fed ad libitum, and it is accepted as the only established antiaging experimental paradigm.
As one of the molecules through which CR improves lifespan extension or delays age-related diseases, initial studies of aging in yeast identified silent information regulator 2 (Sir2), which is a NAD+-dependent deacetylase. Homologues of Sir2 in higher eukaryotic organisms are referred to as sirtuins. SIRT1, the sirtuin that is most closely related to Sir2, is one of seven sirtuins in mammals. The beneficial effects of CR involve the function of SIRT1, which is induced by CR in various tissues. The significance of SIRT1 on the effects of CR has been demonstrated using genetically altered mice. Bordone et al. [4] reported that Sirt1 transgenic mice exhibited a CR-like phenotype, exhibiting reduced levels of blood cholesterol, adipokines, insulin, and fasting glucose and greater glucose tolerance than control mice. However, Sirt1 deficiency in mice fails to extend lifespan under CR [5]. Additionally, a 25% reduction in calorie intake for 6 months in nonobese young adults led to the upregulation of SIRT1 and peroxisome proliferator activated receptor (PPAR)-γ coactivator-1α (PGC-1α) in the skeletal muscle. This effect was accompanied by an increase in mitochondrial function and a decrease in visceral fat mass, insulin resistance, body temperature, metabolic rate, and levels of oxidative stress [6]. Thus, SIRT1 is an important regulator of energy metabolism, and appears to be required for a normal response to CR. Furthermore, recent reports demonstrate that SIRT1 is downregulated in several cells and tissues in insulin-resistant or glucose intolerance states [7-9]. Therefore, under excess energy intake, decreased SIRT1 activity may contribute to the development of obesity-related conditions, including insulin resistance and T2DM. Diet therapy, including CR, is generally necessary for patients with T2DM; however, it is not a simple matter for patients to strictly control their diet over the long term. Therefore, SIRT1 activation, as a CR mimetic, may be a candidate therapeutic target for T2DM.
SIRT1 AND ITS BIOLOGICAL FUNCTION
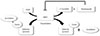
SIRT1 functions as class III histone deacetylases, binding to NAD+ and acetyllysine within protein targets and generating lysine, 2'-O-acetyl-ADP-ribose, and nicotinamide as enzymatic products. Nicotinamide acts as a negative-feedback inhibitor of SIRT1 (Fig. 1).
SIRT1 regulates a wide variety of cellular functions, such as metabolism related to glucose-lipid metabolism, mitochondrial biogenesis, inflammation, autophagy, and circadian rhythms, and others including, stress resistance, apoptosis and chromatin silencing (Table 1) [10]. SIRT1 can act on more than a dozen nonhistone proteins, including transcription factors, transcriptional coregulatory proteins, and histones. SIRT1 participates in the control of systemic metabolism via the regulation of glucose and lipid homeostasis by deacetylating various targets. PGC-1α is an important factor in mitochondrial biogenesis and function and is regulated by an acetylation/deacetylation reaction. The transcription factor forkhead box O1 (FOXO1) is involved in the control of glucose-lipid metabolism and stress resistance. In addition, SIRT1 also regulates components of the circadian clock, such as brain and muscle aryl hydrocarbon receptor nuclear translocator-like 1 (BMAL1) and period 2 (PER2). SIRT1 is associated with lipid metabolism through the activation of nuclear receptors, including PPAR-α, liver X receptor (LXR), and farnesoid X receptor (FXR) and via the negative regulation of sterol regulatory element binding protein (SREBP). Furthermore, SIRT1 deacetylates transcription factors, such as p53, poly-ADP-ribose polymerase-1, hypoxia inducible factors (HIFs)-1α and HIF-2α, nuclear factor (NF)-κB, autophagy-related gene (Atg) 5, Atg7, and light chain 3. These functions mediate stress resistance, apoptosis, hypoxia, inflammatory signaling, and autophagy as physiological responses to environmental toxicity. Thus, the SIRT1 activation may lead to the induction of gene silencing, reduced apoptosis, enhanced mitochondrial biogenesis, the inhibition of inflammation, the regulation of glucose and lipid metabolism and circadian rhythms, the induction of autophagy and adaptations to cellular stress.
SIRT1 AS A THERAPEUTIC PERSPECTIVE IN T2DM
SIRT1 may participate in the control of glucose homeostasis through the following mechanisms: regulating insulin secretion [11] and protecting pancreatic β-cells [12]; improving insulin resistance via the modulation of postinsulin receptor signaling; decreasing inflammation, lipid mobilization, and adiponectin excretion [13]; controlling fatty acid oxidation and mitochondrial biogenesis [14]; and regulating hepatic glucose production and circadian rhythms, skeletal muscle, adipose tissue, monocytes/macrophages, and the liver (Table 2). Therefore, SIRT1 is a promising pharmacological therapeutic target for the treatment of insulin-resistance and subsequent T2DM [15].
Several studies have suggested that SIRT1 participates in the regulation of insulin secretion from pancreatic β-cells. The SIRT1 overexpression in β-cells enhances adenosine triphosphate (ATP) production by repressing uncoupling protein (UCP) 2. This process mediates the uncoupling of ATP synthesis from glucose, and elevated ATP levels lead to cell membrane depolarization and Ca2+-dependent insulin exocytosis. β-Cells in SIRT1-deficient mice, however, produce less ATP in response to glucose than do normal mice. By deacetylating FOXO1, SIRT1 also promotes the activation and transcription of NeuroD and MafA, preserving insulin production and promoting β-cell survival in vivo [11]. Additionally, Lee et al. [12] demonstrated that SIRT1 protects β-cells against various toxic stresses, such as oxidative stress and cytokines, by suppressing NF-κB signaling. In β-cell-specific SIRT1 overexpression (BESTO) mice, increased SIRT1 levels in pancreatic β-cells improve glucose tolerance and enhance insulin secretion in response to glucose [16]. Moreover, SIRT1 activity decreases with age due to decreased systemic NAD+ biosynthesis, resulting in the failure of glucose-sensitive insulin secretion in β-cells. However, the administration of nicotinamide mononucleotide, a metabolite that is important for the maintenance of normal NAD+ biosynthesis, restores glucose-sensitive insulin secretion and improves glucose tolerance in aged BESTO mice [17]. These findings indicate that SIRT1 modulates glucose-sensing ATP production and insulin secretion from β-cells through UCP2, FOXO1, and NAD+ metabolism, resulting in protective effects against various toxic stresses through NF-κB pathway activation.
SIRT1 can directly interact with the insulin signaling pathway through several mechanisms. SIRT1 represses the expression of tyrosine phosphatase1 B, which negatively regulates insulin signaling in skeletal muscle, primarily through dephosphorylation of tyrosine residues on the insulin receptor (IR) and insulin receptor substrate (IRS)-1 [18]. Zhang [19] reported that SIRT1 regulates the insulin-induced tyrosine phosphorylation of IRS-2 through its deacetylation, which affects a crucial step in the insulin signaling pathway. In brief, the insulin-induced tyrosine phosphorylation of the IR and the activation of SIRT1 deacetylase were suggested to be separate events in the insulin signaling pathway. Although IRS-2 is acetylated at the basal state, insulin treatment leads to the tyrosine phosphorylation of the IR, which further recruits IRSs, including IRS-1 and IRS-2, to its kinase domain. The acetylated lysine residues in IRS-2 prevent IR kinase from further phosphorylating the tyrosine residues in IRS-2. Continued phosphorylation of the tyrosine residues in IRS-2 requires the removal of its acetylated lysine residues by insulin-activated SIRT1, and phosphorylated IRS-2 can then serve as an adaptor protein to further transmit insulin signaling to downstream targets, such as Akt [19]. Moreover, Frojdo et al. [9] demonstrated that SIRT1 protein expression was decreased in muscle biopsies and primary myotubes that were derived from subjects with T2DM and that this effect was likely due to posttranscriptional modifications, as no differences in SIRT1 mRNA levels were observed between the controls and type 2 diabetic patients. Moreover, SIRT1 interacts in an insulin-independent manner with the phosphoinositide 3-kinase (PI3K) adapter subunit p85 and modulates insulin signaling at physiological insulin concentrations in skeletal muscle cells [9]. PI3K interacts with IRS following insulin-stimulated tyrosine phosphorylation of IR; insulin signaling can then continue to activate downstream molecules, such as Akt. Therefore, SIRT1 may positively regulate insulin signaling by interacting with PI3K. In addition, the SIRT1 activator resveratrol protects muscle cells, including human primary myotubes, from TNF-α or prolonged hyperinsulinemia-induced insulin resistance. SIRT1 protein can be detected in both nuclear and cytosolic fractions by cell fractionation, and interestingly, nuclear-associated SIRT1 interacts with cytoplasmic proteins, such as IRS-2.
Chronic low grade tissue inflammation is an important etiologic component of insulin resistance and T2DM. Elevated levels of proinflammatory cytokines, such as TNF-α, IL-6, and CRP, in the blood have been detected in individuals with insulin resistance and T2DM. The activation of monocytes in the circulation and adipose tissue has been demonstrated to lead to the release of various inflammatory mediators. Additionally, it has been demonstrated that macrophages residing in adipose tissue may also be a source of inflammatory factors and that these cells may modulate the secretory activity of adipocytes. Tissue macrophages, which are derived from blood monocytes play a central role in both orchestrating and initiating obesity-related tissue inflammatory responses. Moreover, monocytes/macrophages and adipose tissue have reported to exhibit significantly increased binding to NF-κB, the key proinflammatory transcription factor, and an increased levels of intranuclear expression of p65 (Rel A), the major protein component of NF-κB. Thus, the suppression of inflammatory cytokines overproduction in monocytes/macrophages and adipocytes may improve insulin resistance and T2DM. Decreased SIRT1 expression levels in circulating monocytes are correlated with metabolic syndrome, insulin resistance, and glucose intolerance in humans [7]. Moreover, Gillum et al. [8] reported that SIRT1 expression was reduced in adipose tissues of obese males. In addition, mRNA expression of CD14, a macrophage marker, in adipose tissue is negatively correlated with SIRT1 expression. These data indicate that SIRT1 may contribute to the regulation of inflammation in monocytes/macrophages and adipose tissue in humans [8]. Schug et al. [20] also demonstrated that myeloid cell-specific SIRT1 knockout mice that were challenged with a high fat diet displayed high levels of activated macrophages in the liver and adipose tissues, thereby predisposing these animals to the development of systemic insulin resistance and metabolic derangement. SIRT1 physically interacts with the p65 subunit of NF-κB and inhibits transcription by deacetylating p65 at lysine 310, leading to the suppression of inflammatory processes. Yoshizaki et al. [21] provided direct evidence that SIRT1 activation reduced the TNF-α-induced inflammatory response, potentially via the deacetylation of NF-κB (p65) in insulin-resistant adipocytes. Moreover, these authors reported that SIRT1 knockdown in 3T3-L1 adipocytes increased NF-κB (p65) acetylation and enhanced NF-κB binding to target inflammation-related genes promoters [21]. In addition, Yoshizaki et al. [22] reported that SIRT1 represses the activity of the IκB kinase (IKK)-NF-κB signaling pathway, inflammation-related gene expression, and the release of TNF-α following lipopolysaccharide stimulation in macrophages. These authors reported that the pharmacological SIRT1 activator SRT1720 or resveratrol induced various anti-inflammatory activities [22]. Furthermore, the treatment of obese and insulin-resistant Zucker fatty rats with another SIRT1 activator, SRT2379, led to improved glucose tolerance, enhanced systemic insulin sensitivity, and the normalization of tissue markers of inflammation [22]. Additionally, our recent report provided another mechanism with which to explain how SIRT1 inactivation induces inflammation in THP-1 cells. Specifically, SIRT1 inhibition may activate the NF-κB signaling pathway through the phosphorylation of NF-κB (p65) via the dysregulation of autophagy, resulting in the cellular accumulation of p62/Sqstm1 [23]. Moreover, the nutrient-sensing pathway regulates autophagy and involves SIRT1, mammalian target of rapamycin (mTOR) and 5' adenosine monophosphate (AMP)-activated kinase (AMPK). Notably, SIRT1 inactivation resulted in increased mTOR pathway activation and reduced AMPK activation, leading to impaired autophagy [23]. Thus, SIRT1 may attenuate the inflammatory reaction in adipose tissues and monocytes/macrophages and thereby improve insulin resistance and T2DM.
Adipocytes play critical roles in the development of insulin resistance and T2DM given that they can store excess saturated lipids and produce adipokines. PPAR-γ is an essential molecule for the modulation of fatty acid storage and glucose metabolism, and this factor is involved in adipose tissue differentiation. In mature white fat cells, PPAR-γ regulates the induction of genes that are involved in free fatty acid (FFA) uptake and triglyceride synthesis, thereby increasing the lipid storage capacity of the cell [13]. SIRT1 binds to PPAR-γ by docking to the nuclear receptor corepressor and silencing the mediator of retinoid and thyroid hormone receptors, effects that represses the transcription-activating effects of PPAR-γ [13]. Furthermore, SIRT1 overexpression was observed to lead to decreased fat storage and increased lipolysis, resulting in fat mobilization in response to food limitation [13], whereas SIRT1-null mice exhibited a significant reduction in body weight. Additionally, in the adipose tissue of those SIRT1-null mice, the average size of the adipocytes was smaller, the content of the extracellular matrix was lower, adiponectin and leptin were expressed at 60% of the normal level, and adipocyte differentiation was reduced [24]. Moreover, a recent report demonstrated that SIRT1 promotes browning of white fat. SIRT1 deacetylates ligand-bound PPAR-γ on Lys268 and Lys293; therefore, SIRT1 and PPAR-γ coordinately induce the browning of white adipose tissue [25]. These data indicate that SIRT1-dependent PPAR-γ deacetylation regulates energy homeostasis, promoting energy expenditure over energy storage. Therefore, the combination of thiazolidinediones with SIRT1 activator has potential as a therapy for obesity. Adiponectin exerts an antidiabetic effect, and plasma adiponectin levels are decreased in the contexts of obesity, insulin resistance, and T2DM. The administration of adiponectin has been demonstrated to induce glucose-lowering effects and to improve insulin resistance in mice [26]. Moreover, adiponectin-deficient mice exhibit insulin resistance and diabetes [27]. The mechanisms by which adiponectin exerts its insulin-sensitizing effects may be mediated by an increase in fatty acid oxidation via the activation of AMPK and PPAR-α. Additionally, SIRT1 regulates adiponectin expression in adipocytes and FOXO1 forms a transcriptional complex at the mouse adiponectin promoter with CCAAT/enhancer-binding protein α (C/EBPα) [28]. Thus, SIRT1 deacetylates FOXO1 and enhances its interaction with C/EBPα, resulting in the enhanced transcription of the gene that encodes adiponectin in adipocytes. Moreover, a study of muscle adiponectin receptor (adipoR) 1KO mice demonstrated that this protein has a crucial role in the physiological and pathophysiological significance of adiponectin in muscle cells and is involved in the regulation of Ca2+ signaling as well as PGC-1α expression and activation. Adiponectin activates AMPK by biding to adipoR1, thereby activating SIRT1 and deacetylating PGC-1α to improve mitochondrial function, oxidative stress, glucose and lipid metabolism, and exercise endurance [29].
SIRT1 can affect glucose-lipid metabolism and insulin resistance through the modulation of mitochondrial function. The maintenance of energy and nutrient homeostasis during nutrient deprivation is accomplished through an increase in mitochondrial fatty acid oxidation in skeletal muscle. Previous studies have demonstrated a reduced rate of mitochondrial oxidative phosphorylation (OXPHOS) activity and increased intramyocellular lipid accumulation in the skeletal muscle of insulin-resistant patients with type 2 diabetes and elderly individuals [30]. Specifically, these data indicate that defects in mitochondrial function may play an important role in T2DM pathogenesis. An important component that drives this cellular oxidative process in mitochondria is the transcriptional coactivator PGC-1α. PGC-1α activation in skeletal muscle leads to efficient β-oxidation of fatty acids, which is coupled to mitochondrial OXPHOS. In addition, PGC-1α maintains higher numbers of active mitochondria and OXPHOS protein, the levels of which are decreased in T2DM. Through PGC-1α regulation, SIRT1 modulates mitochondrial function and metabolic homoeostasis, increases the consumption of oxygen in muscle fibers and induces the expression of OXPHOS genes and mitochondrial biogenesis. Remarkably, the PGC-1α-induced upregulation of genes that regulate mitochondrial fatty acid utilization was largely prevented by SIRT1 knockdown [14]. Furthermore, SIRT1 can regulate PPAR-α activation through PGC-1α deacetylation, leading to the increased fatty acid oxidation [31]. Thus, SIRT1 activation may improve insulin resistance via accelerated fatty acid oxidation and mitochondrial biogenesis in skeletal muscle. In addition to the effect of increased lipid utilization via PGC-1α-mediated mitochondrial biogenesis, PGC-1α markedly upregulates glucose transporter 4 (GLUT4) expression and glucose transport activity in murine C2C12 myotubes [32]. The effects of PGC-1α on the activation of GLUT4 gene expression are reflected in the increased ability of myocytes to transport glucose, suggesting that the SIRT1-regulated activation of PGC-1α influences insulin sensitization.
The liver plays a central role in glucose and lipid metabolism in response to nutritional and hormonal signals. In a fasted state, the induction of hepatic glucose output and fatty acid oxidation is essential to sustain energetic balance. The production of glucose by the liver is controlled through a complex network of transcriptional regulators. During the early stage of fasting, glucagon induces cyclic AMP (cAMP) response element-binding (CREB) and CREB-regulated transcription coactivator 2 (CRTC2) to drive the expression of gluconeogenesis-related genes that supply the body with the necessary glucose [33]. At the late stage of fasting, SIRT1 is activated and deacetylates CRTC2 to reduce the effects of glucagon [34]. Moreover, at that time, SIRT1 can activate PGC-1α and FOXO1 through a deacetylation reaction, resulting in the induction of gluconeogenesis-related genes. Thus, SIRT1 participates in the regulation of the metabolic switch that controls the shift from the early to the late phase of gluconeogenesis during fasting to maintain glucose homeostasis. Conversely, various reports using animal models have indicated that SIRT1 may have an antidiabetic function. Transgenic mice with moderate SIRT1 overexpression exhibited improved glucose tolerance due to reduced glucose output from the liver [35]. Additionally, Wang et al. [36] also demonstrated that SIRT1 negatively regulates gluconeogenesis. In liver-specific SIRT1-deficient mice, the reduced expression of Rictor, which is a key component of the mTORC2 complex, impaired the Akt-S473 phosphorylation, caused FOXO1-S253 hypophosphorylation, and increased G6pase and Pepck expression to establish chronic hyperglycemia [36]. However, other liver-specific SIRT1 knockout mice exhibit normal glucose levels under both fasting and fed conditions [37]. Moreover, acute SIRT1 knockdown in the mouse liver using an adenovirus system [38] or SIRT1 knockdown in the livers of type 2 diabetic rats using antisense oligonucleotides decreased basal hepatic glucose production and increased hepatic insulin responsiveness to glucose [39]. SIRT1 has also been demonstrated to regulate gluconeogenesis through the deacetylation of signal transducers and activators of transcription (STAT) 3 [40]. STAT3 suppresses gluconeogenesis by inhibiting the transcriptional activity of gluconeogenesis-related gene expression. SIRT1 deacetylates STAT3, resulting in a decrease in STAT3 activity and the subsequent inhibition of gluconeogenesis. Therefore, SIRT1 induces glucose output from the liver in response to fasting via the deacetylation, and thereby inhibition, of STAT3. These results indicate that SIRT1 has a complex role in the regulation of hepatic glucose metabolism under different conditions through the alteration in the expression of gluconeogenesis genes and the modulation of CTRC2, PGC-α, FOXO1, and STAT3 activity.
Dyslipidemia often coincides with T2DM. During fasting or energy limitation, the liver increases lipid utilization and decreases lipid and cholesterol synthesis. Reduced fatty acid oxidation causes hepatic steatosis, which is correlated with insulin resistance. SIRT1 enhances mitochondrial fatty acid oxidation in response to fasting by activating PPAR-α and PGC-1α in the liver. Moreover, SIRT1 regulates SREBP and LXR, both of which are involved in lipid synthesis in the liver: SIRT1 deacetylates and inhibits SREBP-1C activity [41], resulting in decreased lipid synthesis, and deacetylates and positively regulates LXR, contributing to reverse cholesterol transport from peripheral tissues [42]. SIRT1 also activates FXR, which is involved in cholesterol catabolism [43]. In liver-specific SIRT1 knockout mice, the induction of fatty acid oxidation through PPAR-α and PGC-1α was reported to decrease, resulting in increased levels of hepatic FFAs and hepatic steatosis [38,44]. In addition, high fat diet-induced hepatic steatosis was improved in mice with overexpressed SIRT1 [45] and treatment with SIRT1 activators such as resveratrol [41]. Interestingly, recent reports have also indicated that the treatment of obese humans with resveratrol attenuates hepatic fat content and improves insulin resistance [46].
Oxidative stress impairs the insulin signaling pathway and leads to the onset and progression of insulin resistance in T2DM. In hyperglycemia, other metabolites, including FFA and several cytokines, such as TNF-α, induce the overproduction of reactive oxygen species (ROS) by the mitochondria, which are a primary source of ROS. ROS trigger the activation of serine/threonine kinases, such as apoptosis signal-regulating kinase 1, c-jun N-terminal kinase, and IKK, which in turn increase the serine phosphorylation of IRS-1 and decrease the tyrosine phosphorylation of IRS-1. This effect results in insulin resistance and inflammation (oxidative stress linked to inflammation). Thus, reduced mitochondrial oxidative capacity can cause insulin resistance through oxidative stress. PGC-1α deacetylation by SIRT1 mediates mitochondrial biogenesis in addition to the overexpression of antioxidative enzymes, such as Mn-SOD [47], thereby reducing oxidative stress caused by the impaired mitochondria. Moreover, FOXO3a is deacetylated by SIRT1 and translocated to the nucleus, resulting in the upregulated catalase and protection against oxidative stress [48].
The circadian clock, which produces physiological and behavioral rhythms, drives cycles of energy storage and utilization in the anticipation of changes during the day and night, and recent studies have revealed an association between the circadian clock and cellular metabolism. The transcription factors CLOCK and BMAL1 pay a central role in the regulation of circadian gene expression by binding to E-box elements within the promoters of clock-controlled genes (CCGs). Turek et al. [49] demonstrated that homozygous Clock mutant mice exhibited a greatly attenuated diurnal feeding rhythm in addition to hyperphagia and obesity. Moreover, these mice developed a metabolic syndrome that was associated with hyperglycemia, hypoinsulinemia, and hepatic steatosis [49]. Additionally, a high fat diet may disrupt behavioral and molecular circadian rhythms by altering the expression and cycling of clock genes, nuclear receptors and CCGs in the hypothalamus, fat and liver. These findings indicate that nutrient excess may affect the onset and progression of obesity-related diseases, such as diabetes [50]. Moreover, SIRT1 is a key modulator of the circadian clock machinery, and SIRT1 expression or activity both oscillates in a circadian manner and is associated with circadian oscillations in NAD+ levels [51]. The CLOCK-BMAL1 complex interacts with SIRT1 and binds to the promoters of circadian genes, including PER, cryptochrome (CRY), and nicotinamide phosphoribosyltransferase, which encodes the rate-limiting enzyme in NAD+ biosynthesis. Recent reports have demonstrated that SIRT1 participates in the regulation of circadian rhythms via the deacetylation of BMAL1, PER2, and histones H3K9 and H3K14 [51,52]. Acetylated BMAL1 recruits CRY [53], a negative regulator of circadian-controlled gene expression, and promotes the acetylation of PER2, a negative regulator of CLOCK-BMAL1 transcription, thereby enhancing its stability [52]. Thus, SIRT1 links cellular metabolism to the circadian clock in a feedback loop.
SIRT1 ACTIVATING COMPOUNDS
Resveratrol (3,5,4'-trihydroxystilbene), a natural polyphenolic compound that is found in grapes and red wine, is a SIRT1 activator. Numerous reports demonstrate the effects of resveratrol on the improvement of metabolic disorders in ob/ob, db/db, and high fat diet-induced obese mice or Zucker fa/fa rats [15,54]. In addition, resveratrol has exhibited beneficial effects on the longevity and metabolic abnormalities in high fat diet-induced obese mice; however, this compound exhibited no effect on lifespan extension in standard diet-fed mice. In humans, Timmers et al. [46] reported that the administration of oral resveratrol (150 mg/day) to obese male patients for 30 days resulted in CR-like effects, such as improved insulin sensitivity, triglyceride levels, energy expenditure, hepatic lipid accumulation, and the activation of the AMPK/SIRT1 pathway in skeletal muscle. Brasnyo et al. [55] also demonstrated that treatment with resveratrol (10 mg/day) in T2DM patients improves insulin sensitivity and oxidative stress, leading to more efficient insulin signaling via the Akt pathway. However, other recent reports indicate that resveratrol has no effects on metabolism, including insulin resistance. Yoshino et al. [56] demonstrated that oral resveratrol (75 mg/day) supplementation in nonobese and postmenopausal women with normal glucose tolerance does not improve metabolic function, such as insulin sensitivity. Poulsen et al. [57] also reported that high dose of resveratrol (500 mg/day) supplementation in obese men has no effects on the insulin sensitivity, turnover and oxidation of glucose. Thus, the efficacy of resveratrol for metabolism is controversial in humans, and further studies are required.
Resveratrol is not a SIRT1-specific activator, and the mechanism by which resveratrol activates SIRT1 remains unclear. Although resveratrol originally directly can activate SIRT1 allosterically [58], AMPK is required upstream for the activation of SIRT1 by resveratrol. Additionally, Park et al. [59] reported that resveratrol activates SIRT1 through the activation of AMPK via the inhibition of phosphodiesterase 4 and the elevation of cAMP in cells, thereby providing a novel mechanism by which to explain SIRT1 activation by resveratrol. A recent study reported by Price et al. [60] also demonstrated a direct link between SIRT1 and the metabolic benefits of resveratrol. These authors reported that a moderate dose of resveratrol first activated SIRT1 and then induced the deacetylation of liver kinase B 1 and AMPK activation, leading to increased mitochondrial biogenesis and function [60]. Moreover, a high dose of resveratrol may directly activate AMPK, independently of SIRT1.
Synthetic compounds, such as SRT1720 and SRT2379, which are structurally distinct from resveratrol but have potent SIRT1-activating power in vitro have been synthesized by Sirtris Pharmaceuticals. Among these compounds, the treatment of high fat diet-induced obese [21] and ob/ob mice with SRT1720 resulted in an improvement of insulin sensitivity, lower plasma glucose, and increased mitochondrial capacity [54]. In addition, in Zucker fa/fa rats, SRT1720 treatment improved whole glucose homeostasis as evaluated using hyperinsulinemic-euglycemic clamp studies, as well as insulin sensitivity in adipose tissue, skeletal muscle, and liver. Furthermore, Yoshizaki et al. [21,22] also demonstrated the efficacy of the SIRT1 activator SRT2379 against insulin resistance in high fat diet induced obese mice. These authors reported that this effect was related to reduced inflammation in adipocytes and macrophages.
CONCLUSIONS
Over the last decade, our understanding of SIRT1 has expanded from its initial characterization as a single NAD+-dependent class III histone deacetylase that is responsible for longevity in yeast and which is associated with CR. Specifically, it has been found that SIRT1 deacetylates not only histones but also many transcriptional regulators and proteins, thereby modulating diverse biological processes. SIRT1 also may exert antidiabetic effects via the modulation of insulin secretion and improvement of insulin resistance via its regulatory effects on insulin signaling, inflammation, mitochondrial function, and circadian rhythms. Therefore, SIRT1 may be a novel therapeutic target for T2DM.


 XML Download
XML Download