

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Diabetes mellitus is a complex metabolic disorder resulting from progressive impairment of insulin secretion and insulin resistance. Normal pancreatic β-cells exhibit a dramatic response to nutrients and obesity induced insulin resistance via hyper secretion of insulin, which compensates for glucose intolerance. However, in type 2 diabetes, β-cells become unable to sustain a compensatory response, which has a deleterious effect on β-cells [1,2]. Further, there is considerable evidence suggesting that chronic elevation of glucose leads to the generation of reactive oxygen species (ROS), resulting in increased oxidative stress in β-cells [3-5]. As a result, β-cells become worsened with respect to both insulin secretion and action due to their ability to directly damage and oxidize DNA, protein, and lipids. In addition to macromolecular damage, ROS can activate a number of cellular stress-sensitive pathways that have been linked to insulin resistance and decreased insulin secretion [6]. In order to neutralize ROS, cells are equipped with antioxidant defense mechanisms capable of combating oxidative stress. Intriguingly, compared to other tissues, β-cells have a lower abundance of antioxidant defense enzymes such as superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPx) [7,8]. Thus, due to the low antioxidant defense status of islets, excessive ROS lead to oxidative stress during β-cell dysfunction. As such, administration of antioxidant supplements can increase the defense capacity of islet cells to cope with oxidative stress [9-11]. On the other hand, increasing evidence suggests that H2O2 molecules play a role in glucose stimulated insulin secretion (GSIS) [12,13]. Consistent with these reports, induction of endogenous antioxidant capacity in β-cells abrogates ROS signaling and reduces GSIS. Based on these studies, the imbalance between ROS signaling and antioxidant defense can be implicated in diabetes-associated β-cell dysfunction.
SOURCES OF OXIDATIVE STRESS DURING DIABETES
Effect of oxidative stress on hyperglycemia
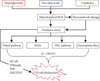
An overwhelming body of evidence indicates that oxidative stress can lead to both cell and tissue injury. Indeed, excess production of such reactive species can be toxic and exert cytostatic effects that cause membrane damage and activate cell death pathways. Healthy pancreatic β-cells exhibit a dramatic response to nutrients and obesity-associated insulin resistance through hypersecretion of insulin in order to maintain energy homeostasis; however, through a complex process that occurs over an extended period of time, β-cells can become unable to sustain a compensatory response, leading to β-cell dysfunction and death [2]. Many studies have suggested that chronic exposure of β-cells to high levels of glucose may contribute to impaired β-cell function [14], resulting in increased glycolytic flux and subsequent production of reducing equivalents leading to production of ROS, including superoxide, hydrogen peroxide, and hydroxyl radicals. Superoxide can subsequently be converted to H2O2 by mitochondrial SOD followed by H2O and oxygen by GPx and catalase. Indeed, several in vitro and in vivo studies have demonstrated that superoxide generation is increased in diabetes. For example, Ihara et al. [15] reported increased oxidative stress markers in Goto-Kakizaki rat β-cells compared with Wistar rat islets [15-17]. In addition, there are several key metabolic pathways activated during hyperglycemia-induced superoxide production, namely, increased polyol pathway activity, increased advanced glycation end products (AGEs) pathway activity, activation of the protein kinase C (PKC) isoform, and increased hexosamine pathway flux. As such, hyperglycemia induced conversion of glucose to sorbitol leads to a concomitant decrease of nicotinamide adenine dinucleotide phosphate (NADPH) and glutathione, which in turn is responsible for the loss of antioxidant equivalents that are more susceptible to elevated intracellular oxidative stress [18]. Under diabetic conditions, an increased flux of glucose through hexosamine biosynthesis pathway (HBP) leads to the formation of uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), an end-product of HBP. Using UDP-GlcNAc as a substrate, O-linked N-acetylglucosamine transferase (OGT) catalyzes the transfer of GlcNAc via O-linkages to specific serine or threonine residues of various target proteins. Indeed, D'Alessandris et al. [19] demonstrated that the increased flux of glucose via HBP impairs insulin-signaling pathways, while other studies have shown that hyperglycemia mediated increases in both UDP-GlcNAc and O-GlcNAcylation leads to both oxidative and endoplasmic reticulum stress, which have been shown to cause chronic inflammation and insulin resistance in other cell types [20]. Furthermore, Kaneto et al. [21] demonstrated increased hydrogen peroxide formation by glucosamine in isolated rat islet cells. In the case of AGEs, glucose reacts with a free amino group to produce adduct formation, which in turn has been shown to interfere with target cell integrity or induce ROS production [22]. Likewise, Jiang et al. [23] demonstrated increased production of hydrogen peroxide with AGEs. Under hyperglycemic conditions, elevated levels of diacylglycerol (DAG) activate PKC, which subsequently increase oxidants such as H2O2 via PKC dependent activation of NADPH oxidase [24]. The above-mentioned pathways clearly show that hyperglycemia-induced overproduction of superoxide by mitochondria is capable of driving multiple pathways [25].
Effect of oxidative stress on lipotoxicity
Recent studies have suggested that elevated glucose along with circulating free fatty acid (FFA) originating from intra-abdominal fat stores is the major culprits of β-cell dysfunction. Indeed, although the exact cause of the metabolic deterioration of β-cells is unknown, several hypotheses have been proposed including mitochondrial dysfunction, oxidative stress, endoplasmic stress, and ceramide formation [26,27]. Several lines of in vitro evidence have indicated that elevated FFA has an adverse effect on mitochondrial function, leading to uncoupling of oxidative phosphorylation and ROS generation [28,29]. Thus, oxidative stress and mitochondrial dysfunction contribute to impaired endogenous antioxidant defenses. In addition, FFA induced formation of ceramide induces generation of ROS and DNA fragmentation [30]. Recent experimental evidence suggests that H2O2 formation in peroxisomes mediates lipotoxicity induced β-cell apoptosis [31]. Specifically, Bindokas et al. [32] quantified superoxide production in islets isolated from Zucker lean fatty (ZLF) and Zucker diabetic fatty (ZDF) rats, and showed that increased superoxide production in ZLF islets was comparable to that of islets of ZDF rats in the presence of glucose. In addition, the resting superoxide content of ZDF rat islets was higher than Zucker lean control islets with perturbed mitochondrial morphology [32].
FFA mediated activation of nuclear factor kappa B contributes to cytokine production and leads to the generation of nitric oxide (NO) through inducible NO synthase (iNOS) expression [33]. In this way, iNOS can result in the overproduction of NO, which in turn can react with superoxide to produce the even more toxic product peroxynitrite. Shimabukuro et al. [34] showed that exposure of prediabetic ZDF rats to FFA upregulates iNOS expression, resulting in a fourfold rise in NO formation and reduced insulin output. As discussed above, generation of ROS and reactive nitrogen species (RNS), as well as the subsequent increase in oxidative stress, may play a central role in the development of diabetes (Fig. 1).
Antioxidant response of islets against oxidative stress
As discussed above, oxidative stress has been associated with β-cell dysfunction in diabetic condition due to their poor antioxidant defense mechanisms. Indeed, there is a delicate balance between oxidants and antioxidants in health and disease, the proper balance of which is essential for cell survival. Thus, redox status is dependent on the degree to which a cell's components exist in an oxidative state, whereby a reducing environment within cells can help to prevent oxidative stress. Such a reducing environment can be maintained by the action of antioxidant enzymes and substances such as glutathione and enzymes such as SOD and catalase, both of which serve to remove ROS. Therefore, induction of endogenous antioxidant enzymes may strengthen islets against detrimental effect of ROS. The main players of intrinsic antioxidant enzymes of islets are SOD, catalase, and GPx, which, compared to liver contents, are 30% and 15% less for SOD and glutathione and catalase, respectively [8,35]. Lortz and Tiedge [36] revealed overexpression of SOD and catalase protects islets against ROS induced impairment of insulin synthesis. In addition, adenoviral mediated over expression GPx has been shown to protect insulin producing INS-1 cells against ROS and RNS insult [37]. Moreover, overexpression of catalase reduces the susceptibility of human and rat pancreatic islets to oxidative stress and preserves insulin secretory capacity [38]. Artificial overexpression of mitochondrial catalase also preferentially protects against oxidative injury and expression of proinflammatory cytokines [39]. In contrast, the ability to overexpress catalase in FVB mice can protect islets against H2O2 and streptozotocin (STZ) toxicity, as well as cytokine toxicity [40] and β-cell specific overexpression of cytoplasmic catalase and methallothionein, which can augment diabetes after cyclophosphamide treatment [41]. In addition, it should be noted that changing the balance of mitochondrial enzymes and increasing production of β-cells can alter susceptibility to dysfunction and development of diabetes.
Many studies have shown that overexpression of UCP2 downregulates levels of ROS [11,12]. Likewise, a study by Kaneto et al. [9] showed that N-acetyl cysteine, along with vitamins C and E, protects metabolically deregulated islets of C57BL/KSJ-db/db mice. Further, alpha-lipoic acid, a dithiol compound and cofactor in mitochondrial energy metabolism, can directly scavenge ROS and RNS in pancreatic islet cells [42]. Administration of alpha-lipoic acid provides a remarkable range of positive therapeutic benefits in nonobese diabetic mice treated with cyclophosphamide [43]. Furthermore, alpha-lipoic acid reduces oxidizing forms of antioxidants including vitamin C and E, as well as elevates GSH levels via its ability to increase cysteine uptake [44]. In addition, a number of reports have shown that alpha lipoic acid improves glucose disposal and reduces body weight in diabetic obese patients [45-47]. Furthermore, Bast and Haenen [48] showed that lipoic acid is capable of reducing an essential component of the mitochondrial respiratory complex cofactor ubiquinone. In support of this observation, exogenous administration of CQ10 blocks cytokine mediated inhibition of GSIS [49].
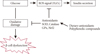
In the present study, a search of novel therapeutic agents identified a set of plant derived flavanoids that exhibited a broad bioactivity spectrum. The identified agents displayed a remarkable array of biochemical and pharmacological characteristics similar to their proposed antioxidant properties, namely, quercetin [50], curcumin [51], ginseng [52], genistein [53], and Epigallocatechin gallate [54] (Fig. 2). The biological properties of the above mentioned flavanoids were identified by limited production of ROS or a scavenging approach based on alternative nonoxidants, including the regulation of cell signaling and gene expression, which comprised vital cellular functions.
CONCLUSIONS
Experimental evidence shows that oxidative stress contributes to β-cell dysfunction and failure in diabetic conditions. Likewise, changes in redox status and depletion of antioxidants occur during oxidative stress induced dysfunction, which suggest the importance of ROS as a signaling molecule in GSIS. Meanwhile, numerous studies have demonstrated that antioxidant therapy potently inhibits ROS generation and eliminates oxidative stress. However, use of these compounds may have limited therapeutic relevance due to their interference with the physiological redox balance. Thus, understanding this complex scenario and determining the proper administration of antioxidants may have a considerable impact on the treatment of β-cell failure during diabetes.
 XML Download
XML Download