

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
AMPK
AMPK is an evolutionary conserved αβγ heterotrimer consisting of an α catalytic subunit and βγ regulatory subunits. In mammals, all subunits are required for a stable complex and full activation of the enzyme [1]. AMPK is activated by low energy status (increased AMP/ADP: ATP) such as during exercise, and regulates metabolic process and energy homeostasis by switching off ATP consuming pathways (fatty acid and cholesterol synthesis) and switching on ATP generating processes (glucose uptake and fatty acid oxidation).
AMPK αβγ heterotrimer expression in skeletal muscle
Skeletal muscle predominately expresses α2β2 complexes with low levels of α1 and β1. AMPK γ1 and γ2 are expressed in multiple tissues [2] whereas γ3 appears to be restricted to skeletal muscle [3-6], where it's association with α and β isoforms differ depending on the fiber composition of the muscle. In mice, γ3 and γ2 AMPK is predominately expressed in fast-twitch glycolytic extensor digitorum longus (EDL) muscle compared to slow-twitch oxidative soleus muscle [4,5] whereas in gastrocnemius muscle, γ1, γ2, and γ3 are evenly expressed [5]. In mice, both AMPK β1 and β2 associate with α2 in EDL whereas β1 AMPK is primarily associated with α2 AMPK complexes in soleus [7]. In humans, α1 and β1 are primarily expressed in slow-twitch oxidative [8] whereas γ3 is predominantly expressed in fast-twitch glycolytic muscle [9]. In human skeletal muscle, the majority of AMPK α2/β2 complexes associate with γ1, whilst the remaining α1/β2 and α2/β2 associate with γ3 [10]. See Table 1 for AMPK isoform heterotrimer distribution in EDL and soleus.
Regulation of AMPK activity: upstream kinases and allosteric regulation
AMPK is inactive unless phosphorylated on the α-subunit activation loop at T172. Both AMP and ADP binding to the γ subunit enhances T172 phosphorylation and suppresses dephosphorylation [11-13]. Phosphorylation is the most potent activator of AMPK increasing activity >100 fold [14]. Upstream kinases, LKB1 and Ca2+/calmodulin-dependent protein kinase kinase (CaMKK) phosphorylate AMPK T172 in mammalian cells. LKB1 is a heterotrimer complex with regulatory proteins STRAD and MO25 [15,16]. In skeletal muscle, studies in two independent models lacking LKB1 have shown that LKB1 is the major AMPK kinase in skeletal muscle [17,18]. Skeletal muscle-specific deletion of LKB1 (LKB1-MKO) from mice results in greatly reduced α2 AMPK T172 phosphorylation following activation by AICAR (a cell-permeable adenosine analog that can be phosphorylated to form 5-aminoimidazoel4-carboxamide-1-d-riborfuronosil-5'monophosphate) or muscle contractions/exercise [17-19]; however, AMPK α1 activity does not appear to be substantially reduced in these models suggesting an alternative upstream kinase may regulate AMPK α1 activity. CaMKK activates AMPK in response to elevated intracellular Ca2+ [20-22]; however, there is a lack of genetic evidence supporting the importance of this kinase in regulating skeletal muscle AMPK T172 phosphorylation.
Insulin resistance
Skeletal muscle constitutes around 45% of lean body mass and is a highly metabolic tissue that is a major contributor to whole-body expenditure. Skeletal muscle is responsible for around 80% of insulin-stimulated glucose disposal [23-25]; therefore, responsiveness to insulin in this tissue is important for regulating whole-body insulin sensitivity. Over the past decade, reduced physical activity levels, due to increasing technologies that promote a sedentary lifestyle and the overconsumption of food, has led to an epidemic of obesity and type 2 diabetes. Obesity and type 2 diabetes are casually linked through their association with skeletal muscle insulin resistance. Insulin resistance is characterized by the reduced responsiveness of target tissues (e.g., skeletal muscle, liver and adipose tissue) to insulin. This results in compensatory hyperinsuliemia, but hyperglycaemia and hyperlipidemia due to the reduced ability of insulin to suppress liver gluconeogenesis and promote glucose uptake in skeletal muscle. Insulin also promotes glucose uptake into adipose tissues and suppresses lipolysis. Therefore, understanding the underlying mechanisms that contribute to the development of insulin resistance as well the pathways involved in improving insulin sensitivity are critical for the development of new strategies (both pharmacological and non-pharmacological such as exercise) that target obesity-induced insulin resistance. Activation of AMPK by pharmacological means have supported a role for AMPK in regulating glucose uptake, fatty acid oxidation, mitochondrial oxidative capacity and insulin sensitivity; therefore, AMPK may be potential target for the treatment of obesity induced insulin resistance given these processes are reduced in skeletal muscle from obese and type 2 diabetic humans and rodents.
AMPK MOUSE MODELS USED TO STUDY SKELETAL MUSCLE METABOLISM
Whole-body α1, α2 [26], β1 [27], β2 [28,29], and γ3 [4] KO and muscle-specific β1β2 KO (β1β2 M-KO) [30] mice as well as transgenic mice overexpressing a kinase dead form of α2 AMPK in skeletal muscle (KD or α2iTg) [31,32] have been generated to help define the role of AMPK in regulating skeletal muscle metabolism. These studies have investigated the role of AMPK in regulating carbohydrate (glucose uptake and glycogen synthesis) and lipid (fatty acid uptake and oxidation) metabolism, and mitochondrial biogenesis and insulin sensitivity at rest and during or following exercise and muscle contractions.
AMPK α
AMPK α2 KO and α2KD mice have reduced AMPK T172 phosphorylation and α2 activity in skeletal muscle, but enhanced or modestly reduced α1 activity, respectively [31,33,34]. In contrast, AMPK α1 KO mice have normal AMPK T172 phosphorylation in skeletal muscle despite reduced α1 activity, showing that α2 AMPK is the predominant isoform contributing to AMPK activity in skeletal muscle [35]. The role of AMPK in insulin sensitivity has been extensively studied in AMPK deficient models fed either a chow or high-fat diet, the latter being considered the most physiological model to replicate human obesity and insulin resistance in rodent studies. AMPK α1 KO and α2KD mice have normal insulin sensitivity on a chow and high-fat diet [33,36]. However, an alternative muscle-specific mouse model on a model FVB background, rather than a C57Bl6 background, overexpressing a muscle-specific KD AMPKα2 B157A mutation (α2iTg), are glucose intolerant and develop exacerbated skeletal muscle insulin resistance when fed a high-fat diet for 30 weeks, which occurs independently of an increase in muscle lipids [37]. In this study, high-fat fed α2iTg mice had reduced IRS1 and Akt protein expression in skeletal muscle compared to WT counterparts, which may have contributed to the reduced skeletal muscle glucose uptake in response to insulin [37]. The FVB strain is known to be resistant to obesity-induced insulin resistance and given that the greater glucose intolerance in α2iTg mice was not evident until after 26 weeks on the high-fat diet, it is unlikely that AMPK is directly involved. Whole-body AMPK α2 KO mice have reduced insulin secretion and whole-body insulin-stimulated glucose utilization due to elevated catecholamine levels and a hyperactive sympathetic nervous system [33]. Basal and insulin-stimulated glucose uptake in isolated muscles is normal in AMPK α2 KO mice, further supporting that insulin resistance was caused by an external defect independent of skeletal muscle AMPK.
AMPK β
Whole-body deletion of both β1 and β2 is embryonic lethal [38]. AMPK β1 KO mice have reduced AMPK activity in liver but not skeletal muscle, are hyperphagic and as a result have lower body weight, adiposity and hepatic lipid accumulation, and are protected from high-fat diet induced insulin resistance [27]. In contrast, AMPK β2 KO mice have reduced AMPK activity in skeletal muscle but not liver or adipose, and have a compensatory increase in AMPK β1 protein expression and maintained α1 expression in skeletal muscle [28]. AMPK β2 KO mice are characterized by reduced exercise capacity and increased susceptibility to develop insulin resistance on a high-fat diet possibly due to greater ceramide accumulation in skeletal muscle [28]. Interestingly, lipid accumulation occurred in AMPK β2 KO mice despite intact fatty acid oxidation rates and increased glycogen utilization in skeletal muscle [28]. An alternative AMPK β2 KO mouse model has recently been generated and these mice have a similar phenotype the β2 KO model reported by Steinberg et al. [28], but in addition, show that AMPK plays an important role in the adaptive response to fasting whereby AMPK β2 KO mice have reduced phosphorylation of S6K and the AMPK substrate ULK1, which is involved in autophagy [29,39].
We have recently generated muscle-specific AMPK β1β2 double KO mice (β1β2M-KO) to alleviate concerns related to residual AMPK activity due to remaining subunit homologs being present or upregulated in previous AMPK deficient models (α2 KO, α2KD, β2 KO, γ3 KO). As anticipated, AMPK β1β2M-KO mice have undetectable levels of AMPK activity at rest or during exercise [30]. Their phenotype is more dramatic compared to whole-body AMPK β2 KO mice [28] and is characterized by dramatically reduced voluntary wheel activity (75%), maximal exercise capacity (95%), mitochondrial content (30%) and exercise-mediated glucose uptake in skeletal muscle (55% to 70%) [30]. Interestingly, young (9 to 12 weeks) AMPK β1β2M-KO mice have similar whole-body weight and adiposity, and consistent with this, whole-body and skeletal muscle specific insulin sensitivity is similar when compared to wild-type littermates [30]. These studies highlight an important role for AMPK in regulating exercise capacity, mitochondrial content and glucose uptake during exercise, but not insulin sensitivity.
AMPK γ
The γ1 subunit is ubiquitously expressed similar to γ2; however, γ2 expression is predominant in heart [2,3]. In contrast, γ3 appears to be expressed specifically in skeletal muscle [3-6]. Studies from both whole-body γ3 KO and transgenic γ3 overexpressing or gain-of-function mice have revealed an important role for the γ3 subunit in regulating glucose uptake, glycogen synthesis, fatty acid oxidation and mitochondrial biogenesis in skeletal muscle [40]. Whole-body γ3 KO mice have reduced AMPK activity at rest, during exercise and in response to fasting, but normal insulin sensitivity under basal conditions and glucose uptake during exercise [4,41]. Interestingly, gain-of-function mutations in the γ subunits result in glycogen storage disease in humans [42-44] and pigs [45]. AMPK γ3 R225Q mutation is the most studied mutation and this mutation is homologous to R225Q in Hampshire pigs, as well as AMPK γ2 R200Q in humans, which is associated with Wolff-Parkinson-White syndrome [46,47]. Interestingly, transgenic mice harboring the R225Q mutation in AMPK γ3 subunit have higher AMPK activity and glycogen levels and are protected from diet-induced insulin resistance [4], which is a likely effect of increased fatty acid oxidation and subsequent reduction in skeletal muscle lipids. Mice with an AMPK γ3 R225Q mutation are also fatigue resistant during exercise whereas AMPK γ3 null mice are prone to fatigue during exercise and have impaired glycogen re-synthesis following exercise (see section AMPK and glycogen synthesis) [4,48]. Whilst γ3 appears to be predominantly expressed in glycolytic muscles, training reduces γ3 expression in humans [10,49], which may be an adaptive response to enhanced oxidative capacity (preference for lipid oxidation). These studies suggest that targeting AMPK may enhance exercise performance and be protective against insulin resistance disease development in obesity.
AMPK AND EXERCISE
An important feature of skeletal muscle is its ability increase ATP turnover (>100 fold) during exercise in order to provide energy for contracting muscles [50]. Under such conditions, AMP and ADP levels are rapidly increased in an intensity-dependent manner and ATP levels decline slightly. AMPK is a sensor of cellular energy status that is activated by AMP and ADP. Consistent with this, AMPK is activated during exercise in both rodents and humans [51-60], in an intensity-dependent manner [57,59-61]. Both AMPK α1 and α2 are activated during exercise; however, α1 activity appears to be increased during high intensity muscle contractions, which are equivalent to >100% maximal oxygen consumption (VO2 Max; measure of maximal exercise capacity) [59,62,63]. In contrast, α2 activity is increased at low intensity exercise starting at 40% VO2 Max and increases progressively with exercise intensity.
Exercise can also regulate glucose uptake by an insulin-independent mechanism involving AMPK [30]. Importantly, GLUT4 translocation during exercise is normal in skeletal muscle of type 2 diabetics [64], and exercise also improves insulin sensitivity. For these reasons, exercise is often advised for both the prevention and treatment of insulin resistance. Therefore, understanding the underlying mechanisms regulating glucose uptake is an important area of research.
REGULATION OF GLUCOSE UPTAKE
Insulin signaling
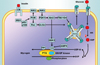
The first step in understanding how glucose uptake is regulated during exercise and how insulin sensitivity is enhanced following exercise is to understand how insulin regulates glucose uptake in normal healthy individuals. In brief, insulin promotes glucose uptake through a signaling cascade consisting of a number of spatially distinct phosphorylation events that result in moving glucose transporters (GLUT4) to the plasma membrane, which upregulates glucose transport into the cell (Fig. 1). Specifically, insulin mediates glucose uptake by binding to its tyrosine kinase receptor on the outside of the cell, causing further activation/phosphorylation of the IRS proteins-1 and 2 inside the cell. Activation of IRS proteins trigger the activation of class I phosphatidlyinositol-3-kinase (PI3K) and subsequent interaction with p110 catalytic and p85 regulatory subunits via increased interaction with SH2 domains, which allows phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2) to form phosphatidylinositol 3,4,5-triphosphate (PIP3) at the plasma membrane. This promotes Akt recruitment to the plasma membrane via PH domain interaction with PIP3, which induces a conformational change in Akt that allows phosphorylation at T308 S473 and activation [65]. Interaction with the activating kinase phosphoinositide-dependent kinase (PDK) phosphorylates T308 in the activation loop and leads to partial activation of Akt/protein kinase B (PKB) [66]. The complete activation of Akt requires phosphorylation of Akt at the hydrophobic C-terminal domain S473 residue by mammalian target of rapamycin complex 2 (mTORC2) [67]. Akt phosphorylation of TBC1 (tre-2/USP6, BUB2, cdc16) domain family, member 4 (TBC1D4) (formerly known as Akt substrate of 160 kDa; AS160) at T642 and phospho S/T Akt (PAS) sites, with the (R/K)X(R/K)XXS*/T* recognition motif, is important for GLUT4 translocation in response to insulin. Phosphorylation of TBC1D4 increases binding to 14-3-3 proteins [68-70], and this has been proposed to inhibit RabGAP function towards downstream Rabs; thereby, promoting GTP loading and activation of Rabs and the release of glucose transporter GLUT4 from intracellular compartments to the plasma membrane for glucose uptake [69,71-73].
TBC1D4
Correlation studies suggest that T642 (human and T649 in mouse) is the predominant Akt site on TBC1D4 recognized by the PAS antibody [74]. In mice, whole-body mutation of S642 (S649 in mouse) to alanine (TBC1D4 T649A KI) reduces whole-body glucose and insulin intolerance and impairs insulin-stimulated glucose uptake and GLUT4 translocation in muscle [75]. Similarly, overexpression of TBC1D4 by mutation of four predominant Akt phosphorylation sites (S318, S588, T642, and S751) (4P mutant) in mouse skeletal muscle inhibits insulin-stimulated glucose uptake in vivo by ~50% [76]. Interestingly, this defect is rescued by introduction of a mutant inactive Rab/GAP (R/K), suggesting that this domain is essential for insulin-stimulated glucose uptake. In human skeletal muscle, Treebak et al. [77] have shown that using site-specific antibodies against S318, S341, S588, and S751, TBC1D4 phosphorylation is increased following insulin stimulation during a hyperinsulinemic clamp. These studies support that phosphorylation of TBC1D4 and subsequent 14-3-3 binding and deactivation of this RabGAP is important for insulin-stimulated glucose uptake. It has been proposed that AMPK may phosphorylate TBC1D4 at T642 and promote 14-3-3 binding; however, this has not been confirmed in studies using various cell lines (L6 myotubes, 3T3L1 adipocytes) and cell free assays [70,78-80]. TBC1D4 may also play a role in the insulin sensitizing effects of exercise [77], as exercise training has been shown to restore TBC1D4 phosphorylation in skeletal muscle from type 2 diabetics [81].
Akt2 is essential for TBC1D4 T642 and PAS phosphorylation in response to insulin [76]. Akt2 KO mice and siRNA targeted silencing of IRS1 and Akt2 in human myotubes have blunted insulin-stimulated TBC1D4 phosphorylation compared to their homologous counterparts IRS2 and Akt1 and 3 [68,82]. Reduced IRS1 and Akt phosphorylation has previously been considered an important component leading to reduced insulin-stimulated glucose uptake that is associated with insulin resistance. However, recent evidence suggests that low levels of Akt activation are sufficient for maximal GLUT4 translocation in response to insulin [83], and modest reductions in IRS1 do not necessarily translate into impaired insulin action in skeletal muscle, which has been highlighted in IRS1 heterozygous mice [84] and shRNA mediated silencing of IRS1 in vivo that display normal glucose uptake in response to insulin [85]. A further explanation for these observations whereby insulin signaling is normal despite reduced insulin-stimulated glucose uptake is that insulin-stimulated glucose uptake can also occur independently of PKB/Akt signaling [86]. This process still requires PI3K but involves activation of Rac1, a small GTPase protein from the Rho family that promotes glucose uptake by association with actin; thereby promoting actin remodeling [87].
GLUT4 vesicles move along cytoskeletal elements that are directed by GTP-bound (active) Rab-proteins; small G proteins required for membrane trafficking [69]. Actin remodeling is a dynamic multistep process that occurs at the cell surface, and changes in the spatial and temporal arrangement of actin are considered important for providing a scaffold for the transmission of signaling from insulin receptor to GLUT4. Whilst the regulation of actin by Rac1 and the interaction between actin and GLUT4 vesicles in regulating insulin-stimulated glucose uptake has not been extensively studied compared to the Akt/PKB pathway, it is known that that Rac1 is rapidly activated (GTP loaded) in response to insulin in both adipose and muscle cells, and a dominant-negative Rac mutant unable to bind GTP prevents insulin-induced actin remodeling [88]. Evidence that the Rac/actin and PKB are independent pathways both regulating glucose uptake in response to insulin is supported by studies showing that 1) siRNA knockdown of Rac1 in L6 myotubes reduces insulin-stimulated GLUT4 translocation without altering Akt phosphorylation [89]; 2) inhibition of P13K with wortmannin prevents actin remodeling [90] and in contrast, 3) overexpression of a dominant negative Akt mutant does not alter actin remodeling in L6 myotubes [91]. Few studies have investigated the involvement of the Rac/actin pathway in insulin resistance; however, ceramides, which are bioactive lipids that are elevated in obese skeletal muscle and reduce insulin-stimulated glucose uptake, reduce insulin-stimulated actin remodeling and GLUT4 translocation in L6 myotubes [89]. PKC has also been implicated in regulating GLUT4 trafficking to the plasma membrane in response to insulin; however, PKC appears to lie downstream of Rac [92].
TBC1D1
Recently, TBC1D1, a closely related paralog of TBC1D4, has also been identified as an Akt substrate that may play a critical role in regulating GLUT4 trafficking events. TBC1D1 is a candidate gene for the development of severe familial obesity in females, due to the presence of an R125W coding variant that has been identified in 4p15-14-linked obesity pedigrees from American and French populations [93,94]. It is not currently known how the R125W mutation increases susceptibility to obesity; however, it's localization in the phosphotyrosine binding domain of the N-terminus may affect TBC1D1 function [93]. Expression of the R125W mutation in mice reduces insulin-stimulated glucose uptake in skeletal muscle [95]. The C-terminus of TBC1D1 includes a TBC (Tre-2/BUB2/Cdc16) domain that contains Akt, AMPK, and PKA phosphorylation sites thought to be important for regulating Rab GAP activity and association with Rab GPTases; however, the R125W mutation does not affect TBC1D1 PAS phosphorylation [93,95], which suggests that they have distinct functions in regulating glucose uptake. Silencing TBC1D1 in L6 muscle cells by siRNA increases basal and insulin-stimulated GLUT4 translocation [71], and overexpression of TBC1D1 in 3T3-L1 adipocytes with low endogenous TBC1D1 expression inhibits insulin-stimulated GLUT4 translocation, which is reversed by expression of a mutated form of TBC1D1 whereby the GAP domain has been inactivated [96]. Recombinant congenic mice lacking TBC1D1 are protected against diet-induced obesity, which is likely due to increased fatty acid uptake and oxidation in skeletal muscle [97]. These studies support a role for TBC1D1 in regulating glucose uptake.
TBC1D1 and TBC1D4 share 47% sequence identity; however, their GAP domains are 79% identical and 91% similar [96]. Like TBC1D4, TBC1D1 exerts a regulatory role in glucose uptake and metabolism through regulation of its GAP activity. In contrast to TBC1D4, TBC1D1 is only phosphorylated on one of the five standard Akt motifs (T590) and one partial one (S501), which corresponds to T649 and S577 of TBC1D4 in mouse, respectively. The PAS antibody recognizes T590 on TBC1D1, which is the equivalent T642 site on TBC1D4. It has been predicted that this site may also be an AMPK site [74]. Unlike TBC1D4, 14-3-3 binding to TBC1D1 does not occur upon insulin stimulation [70]. TBC1D4 and TBC1D1 also differ in their expression in tissues, whereby TBC1D1 is predominately expressed in skeletal muscle with little in adipose and none in heart whereas TBC1D4 is ubiquitously expressed [98]. Of note, TBC1D4 is predominately expressed in oxidative soleus muscle, whereas TBC1D1 is more highly expressed in glycolytic muscle such as tibialis anterior and extensor digitorum longus [98]. The predominant expression of TBC1D4 in oxidative soleus muscle compared to TBC1D1 may be related to its predominant role in regulating insulin-stimulated glucose uptake, which would be consistent with greater insulin binding capacity of oxidative muscles.
A number of additional Akt phosphorylation sites on TBC1D1 have also been identified (see [99]), and mutation some of these sites (T499, S489, S501, and T590) to alanine in C2C12 myotubes reduces GLUT4 translocation to the cell surface in response to insulin; however, S501 and T590 appear to be the most predominant regulators of TBC1D1 GAP activity controlling this process [99]. Interestingly, the combination of AICAR with insulin is additive in relieving TBC1D1 inhibition on GLUT4 translocation in both wild-type and mutant cells compared to each treatment alone, suggesting a possible role for AMPK in the regulation of TBC1D1 activity [99]. In contrast to the above findings, An et al. [95] has shown that overexpression of TBC1D1 by mutation of four potential AMPK/Akt phosphorylation sites (4P) (S231, T499, T590, S621) in skeletal muscle does not impair insulin-stimulated glucose uptake in vivo, suggesting that alternative AMPK sites may be important or AMPK does not play a role in regulating insulin-stimulated glucose uptake via TBC1D1. In support of this, insulin also does not increase AMPK T172 or ACC S79/212 phosphorylation [74] and AMPK α2, β2 and γ3 null and α2KD and β1β2 M-KO mice have normal insulin-stimulated glucose uptake in muscle [26,28,30,36,100] despite blunted TBC1D1 PAS and T590 phosphorylation in response to insulin [30,74].
Under basal conditions, a role for AMPK in regulating TBC1D1 has been suggested given that 1) the Akt inhibitor wortmannin does not alter basal TBC1D1 phosphorylation [74,101]; 2) Akt2 KO mice have normal basal TBC1D1 phosphorylation [102]; and 3) AMPK α2 KD, α2 KO α2iTg and β1β2 M-KO mice have reduced basal TBC1D1 phosphorylation [30,74,102]. Collectively, these studies suggest that whilst Akt is important for mediating insulin-induced TBC1D1 phosphorylation, AMPK activity is important for maintaining basal levels of T590 phosphorylation. In further support of this, it has been shown that TBC1D1 co-localizes with GLUT4 in response to A-769662 in L6 myotubes [70]. Based on these findings, AMPK may be important for TBC1D1 localization and priming in order to allow phosphorylation by Akt following insulin stimulation. Further investigation on TBC1D1 localization in muscle-specific AMPK β1β2-KO myotubes in response to different stimuli would be of interest for determining this.
Exercise and glucose uptake
Like insulin, exercise increases the rate of glucose uptake into contracting skeletal muscles, by a process regulated by GLUT4 translocation to the plasma membrane and transverse tubules. Interestingly, insulin and exercise/contractions regulate glucose uptake in skeletal muscle by distinct mechanisms (Fig. 2). This revelation came from the following original observations:
1) PI3 kinase inhibitor wortmannin inhibits insulin but not contraction-stimulated glucose uptake. Contraction does not promote IRS-1 autophosphorylation or increase PI3 kinase activity [103-106].
TBC1D4
The involvement of TBC1D4 in regulating glucose uptake in response to contraction has been extensively summarized (see review [73]). In brief, evidence that TBC1D4 is involved in contraction-mediated glucose uptake came from the following studies:
1) Rat epitrochlearis muscle contracted in vitro in the absence of insulin increased TBC1D4 phosphorylation [115].
2) Contraction and insulin additively increased TBC1D4 phosphorylation [115].
3) TBC1D4 4P mutant has impaired contraction-mediated glucose uptake [68].
AMPK rather than Akt has been proposed to be a possible candidate regulating TBC1D4 activity in response to exercise and muscle contractions given 1) Akt phosphorylation is not increased by exercise; 2) AICAR stimulates glucose uptake independently of insulin; 3) AICAR increases phosphorylation of TBC1D4 at PAS motifs in both rat [115] and mouse skeletal muscle [76]; and 4) glucose uptake is impaired in the TBC1D4 4P mutant in response to AICAR and contraction [76]. The binding of Ca2+/calmodulin to the Ca2+/calmodulin binding domain (CBD) of TBC1D4 also regulates glucose uptake in response to exercise but not insulin. However, combining the 4P mutant with a CBD mutant results in similar reductions in glucose uptake when compared to the 4P and CBD mutants alone [116], suggesting that each is necessary but not sufficient for regulating this process.
Alternative studies provide evidence that PAS-TBC1D4 phosphorylation is not essential for contraction-mediated glucose uptake given that increases in phosphorylation do not appear at the onset of exercise when glucose uptake is rapidly increased (reviewed in [73]). Consistent with this, TBC1D4 T649A KI mice do not have impaired AICAR and contraction mediated glucose uptake, suggesting that the other three sites (S325, S595, and S758) may also be important for regulating TBC1D4 activity and glucose uptake during exercise [78]. Of note, TBC1D4 PAS phosphorylation is not increased with contraction in TBC1D4 T649A KI mice, supporting that TBC1D4 PAS antibody primarily recognizes the T649 site. Worth mentioning is that the S595 site has also been shown to be a potential AMPK site [80], and indeed, its phosphorylation was increased upon in situ contraction in muscle extracts from both wild-type and TBC1D4 T649A KI mice [78]. Further studies in alternative knockin mutant mice may be important for further defining the role of TBC1D4 in regulating glucose uptake in response to exercise and contraction.
TBC1D1
Like TBC1D4, TBC1D1 is phosphorylated in response to AICAR and muscle contractions/exercise [74,95,101,102,117]. In response to muscle contractions, increases in TBC1D1 phosphorylation are important for 14-3-3 binding, inhibition of GAP domain function and subsequent increases in glucose uptake [95,102]. This is in contrast to TBC1D4 whereby 14-3-3 binding is not considered important for regulating glucose uptake during exercise [78]. AMPK phosphorylates multiple sites on TBC1D1 (S231, S499, T590, S660, S700), and muscle contractions/exercise predominately induces TBC1D1 phosphorylation on S231 in both humans and mice [74,102,117]. TBC1D1 T590 is also predominately phosphorylated in response to muscle contractions [74]; however, this has not been observed in all studies [102]. Interestingly, two separate studies inactivating 4 predicated TBC1D1 phosphorylation sites (4P mutant) results in 25% to 30% reduction in contraction-mediated glucose uptake [95,102]. An et al. [95] mutated conserved Akt site (T596) and 3 predicted AMPK sites (S231, T499, S621) whereas Vichaiwong et al. [102] mutated 4 predicted AMPK sites (S231, S499, S660, S700).
TBC1D1 expression is normal in skeletal muscle from type 2 diabetics [99], suggesting a possible explanation for normal glucose uptake during exercise in these subjects. In contrast, TBC1D1 expression is reduced by ~50% in skeletal muscle of AMPK α2 KO, α2 KD, α2iTg and β1β2 M-KO mice [30,74,102,117], and this may be compensatory for the lack of phosphorylation and inhibition by AMPK, or alternatively, reduced muscle glucose uptake in AMPK β1β2 M-KO mice. TBC1D1 PAS phosphorylation is slightly increased in α2iTg and α2KO mice but unresponsive in AMPK KD and β1β2 M-KO mice following contractions [30,74], suggesting that AMPK is important for PAS phosphorylation in response to contraction. However, the effect of AMPK on regulating glucose uptake during exercise is less clear. AMPK α2 KO and α2 KD and α2iTg mice have normal or only modest reductions in glucose uptake during contractions [4,26,31,32,118-120]. It appears that only when AMPK activity is completely abolished, as in β1β2 M-KO mice, that more dramatic reductions in glucose uptake are observed [30], which are similar to that observed in LKB1-MKO mice [18]. Although TBC1D1 expression or PAS phosphorylation was not assessed in skeletal muscle from β2 or γ3 KO mice, contraction-mediated glucose uptake was also normal [4,28], suggesting that the remaining subunit isoforms (likely contributing to maintained or elevated α1 AMPK activity) was sufficient to allow for normal glucose uptake. In addition to AMPK, the AMPK-related kinase SNARK has also been shown to be important for glucose uptake during contraction of mouse skeletal muscle [121]. Collectively, these studies support a role for AMPK and possibly AMPK-related kinases in regulating glucose uptake during exercise and muscle contractions. Alternative unidentified pathways may also exist and further studies are required to fully define the underlying mechanisms regulating GLUT4 translocation in response to exercise.
EXERCISE AND INSULIN SENSITIVITY
Exercise enhances insulin-stimulated glucose uptake, an effect that can persist for hours following an acute bout of exercise. The underlying mechanisms responsible for exercise-induced improvements in insulin sensitivity are not currently known; however, a number of possible candidates have been suggested (including TBC1D4, AMPK, interleukin 6 [IL-6], adiponectin, glycogen synthesis/levels, fiber-type/oxidative capacity). Arias et al. [122], was one of the first to show that TBC1D4 PAS phosphorylation was elevated in rat skeletal muscle following an acute bout of exercise, an effect that persisted for 3 to 4 hours postexercise. These studies lead to the idea that TBC1D4 may be involved in the insulin sensitizing effects of exercise. Since then, studies comparing insulin sensitivity following both in vivo exercise and in vitro contraction of isolated muscles have been performed. These studies have found that whilst TBC1D4 T642 and PAS phosphorylation is increased immediately after exercise and 3 hours postexercise consistent with enhanced insulin-stimulated glucose uptake; following in vitro contractions TBC1D4 phosphorylation had returned to resting levels 3 hours postexercise despite having an additive effect on glucose uptake in response to insulin [123]. It was proposed that circulating factors such as IL-6 may be involved in postinsulin sensitizing effects of exercise; however, TBC1D4 phosphorylation still returned to resting levels even when serum was added to the incubation media or not. These studies indicate that whilst circulating factors may also play a role in the insulin sensitizing effects of in vivo exercise (see IL-6 and adiponectin section); unknown TBC1D4-independent mechanisms are also involved following in vitro contractions. Studies in humans have also revealed similar findings, whereby 60 minutes of leg extension exercise (80% of maximal exercise capacity) increases TBC1D4 phosphorylation on specific sites (S318, S341, S588, and S751) but not PAS or T642 phosphorylation 4 hours post exercise [77]. In this study, Treebak et al. [77] also found that 14-3-3 binding to TBC1D4 in skeletal muscle was not further enhanced by leg exercise when compared to insulin alone despite increased insulin-stimulated glucose uptake following exercise, suggesting that insulin sensitivity following exercise may not involve 14-3-3 binding to TBC1D4. Similarly, Howlett et al. [124] found that although 3 hours of cycling increased 14-3-3 binding to TBC1D4 immediately following exercise, this had returned to baseline despite maintained TBC1D4 phosphorylation 3 hours postexercise. Whilst these studies support a role for TBC1D4 in insulin sensitivity following exercise, it also suggests that alternative mechanisms are also involved.
Unlike TBC1D4, TBC1D1 requires 14-3-3 binding in response to contraction and AICAR but not insulin in both human and rodent skeletal muscle [74,117]. TBC1D1 phosphorylation is enhanced immediately following exercise but returns to basal levels 3-4 and 27 hours post-exercise in rats, suggesting that the involvement of TBC1D1 in the insulin sensitizing effects of exercise are different from that of TBC1D4. Both acute exercise and AICAR activate AMPK and increase insulin sensitivity. These findings have led to the idea that AMPK may be involved in the insulin sensitizing effects of exercise. Phosphorylation of TBC1D1 at S237 appears to be the predominant site phosphorylated by AMPK; however, TBC1D1 PAS phosphorylation does not appear to correlate well with S237 phosphorylation [74]. No studies have assessed the TBC1D1 phosphorylation at specific sites following exercise or investigated insulin sensitivity in TBC1D1 4P mutant, but these studies would provide valuable evidence about the involvement of AMPK and TBC1D1 in the insulin sensitizing effects of exercise.
AMPK AND GLYCOGEN SYNTHESIS
Glycogen synthesis is increased following exercise and during insulin stimulation, and insulin-stimulated glycogen synthesis is enhanced following exercise. Contraction and insulin promote glycogen synthesis by distinct mechanisms but both involve multisite dephosphorylation and activation of glycogen synthase (GS). In the case of insulin-stimulated glycogen synthesis, insulin signaling thru Akt inactivates GSK3 by phosphorylation at S9 and 21, and this in turn activates GS. In contrast, contraction requires protein phosphatase 1 (PP1) regulatory unit R(GL) to activate GS [125]. GS is phosphorylated on at least nine sites and insulin and contraction dephosphorylates both C- and N terminal sites [126]. Insulin-stimulated glucose transport enhances cellular G6P, which in turn promotes glycogen synthesis. Mutation of the G6P allosteric binding site on GS R582 to alanine (GS R582 KI) in mice reduces glycogen synthesis in response to insulin, suggesting that the G6P allosteric control of GS is also important in controlling glycogen synthesis [127]. Similarly, mutation of two Akt phosphorylation sites on GSK3 (S9 and S21) to alanine prevents inhibition of GSK3 and results in impaired insulin-stimulated glycogen synthesis [128]. In regards to AMPK, the AMPK β subunits contain a central domain, referred to as the carbohydrate-binding motif (CBM) or glycogen-binding domain that allows AMPK to bind to glycogen [129]. This domain is highly conserved between isoforms and across species, ranging from mammals, fruitfly (Drosophila melanogaster), worm (Caenorhabditis elegans), yeast (Saccharomyces cerevisiae), and plants [129]. The CBM is closely related to isoamylase domains found in glycogen and starch branching enzymes [129]. Recently, mutations in the CBM have been shown to prevent glycogen binding and reduce total AMPK activity in cell-free assays [130] and in vivo [131]. Glycogen can also inhibit AMPK, suggesting that AMPK may regulate cellular energy status through glycogen sensing [130].
AMPK regulates glycogen synthesis through phosphorylation of glycogen synthase (GS) at site 2 (S7) [132]. This inhibits enzyme activity in isolated muscles treated with AICAR [133-135]. In liver, GS inactivation by AMPK activators (AICAR and A769662) occurs at S7 phosphorylation site on GS2, which is the liver-specific isoform [136]. Interestingly, AMPK activation by AICAR promotes glucose uptake and increases G6P, which can be used for glycogen synthesis or metabolized through glycolysis to produce ATP [127]. Recently, acute treatment with AICAR was shown to promote glycogen synthesis in skeletal muscle through a G6P-GS dependent pathway; an effect that is blunted in GS R582A KI mice [137]. These studies suggest that GS inhibition by AMPK can be overridden by G6P. LKB1-MKO and AMPK α2 KO, β2 KO and α2 KD mice have lower starting glycogen levels in skeletal muscle [17,138-140], supporting a role for AMPK in glycogen synthesis.
Glycogen synthesis following exercise
It was previously thought that reduced glycogen synthesis in skeletal muscle from type 2 diabetics contributes to reduced insulin-stimulated glucose uptake [141]. Exercise promotes glycogen resynthesis and this may be important for improving insulin sensitivity postexercise. Funai et al. [142] found that refeeding rather than fasting following exercise reduced the enhanced effects of insulin on glucose uptake as well as TBC1D4 T642 and PAS phosphorylation. These studies suggests that whilst increases in insulin-stimulated glucose uptake following exercise may be to allow for enhanced glycogen synthesis, nutritional state can also influence the magnitude of improvements in insulin sensitivity observed following an acute bout of exercise. It has been proposed that glycogen depletion and AMPK activation may play a role in insulin sensitizing effects of exercise given AMPK γ3 KO and α2 KD mice have impaired glycogen resynthesis following exercise [4,31]. However, alternative rodent studies investigating the relationship between AMPK activation, glycogen synthesis and insulin sensitivity under different contraction (contraction length, frequency, and time) and exercise intensity (high [HIT] vs. low [LIT]) protocols have found inconsistent observations. Koshinaka et al. [143] found that whilst AMPK activation increased with exercise intensity, there were not greater improvements in insulin sensitivity when HIT was compared to LIT exercise groups. Similarly, Kim et al. [144] showed that whilst the different contraction protocols altered the level of AMPK activation, there were no differences in the level of improvement in insulin sensitivity. Furthermore, improvements in insulin sensitivity were not consistent with AMPK activation and glycogen depletion [144]. These studies suggest that whilst AMPK may be involved in insulin sensitizing effects of exercise, alternative pathways and measures other than glycogen levels and AMPK activity should be considered when determining the level of exercise-mediated improvements in insulin sensitivity.
ALTERNATIVE MECHANISMS LINKING AMPK TO INSULIN SENSITIVITY
IR and IRS
Exercise improves insulin sensitivity and it was originally proposed that this may involve the direct interaction of AMPK with IRS1 [145]. It has been reported that AMPK rapidly phosphorylates IRS1 on S789 in vitro and treatment with AICAR increases IRS1 S789 phosphorylation in mouse C2C12 myotubes, suggesting a possible link between the potentiating effects of exercise on insulin signaling [145]. Alternative studies show that IRS1 S789 phosphorylation is negatively associated with insulin signaling [146-149]. Instead, activation of AMPK by glucose deprivation or AICAR promotes tyrosine phosphorylation of IR and IRS-1 as well as Akt T308 and S473 phosphorylation [146]. Whilst AMPK is not a tyrosine kinase, it is hypothesized that S/T phosphorylation of IR and IRS-1 by AMPK promotes allosteric activation by tyrosine kinases; however, further studies are required to confirm this [146].
mTOR
An alternative mechanism that may explain the potential insulin sensitizing effects of exercise involves AMPK inhibition of the mammalian target of rapamycin/p70 ribosomal S6 kinase (mTOR/p70 S6K) signaling pathway [150]. Obese and type 2 diabetic rodents have increased mTOR/S6K signaling and this may contribute to insulin resistance [151-153]. S6K phosphorylation of IRS1 at S1101 is increased in genetic and diet-induced obese mice and has shown to induce insulin resistance [154]. Inhibition of mTOR/p70 S6K with rapamycin restores insulin sensitivity caused by amino acids in human skeletal muscle [155,156]. Endurance training has been shown to inhibit mTOR/p70 S6K signaling and reduce IRS1 serine phosphorylation in rat skeletal muscle [157], and recent studies have demonstrated that AMPK is involved in the suppression of mTOR signaling [39]. For example treatment with AICAR inhibits S6K (mTOR effector) activity in a range of mammalian cells [150]. Similarly, incubation with AICAR in C2C12 myotubes transfected with an adenovirus of a constitutively active (truncated form of AMPKα1) or KD form of AMPKα revealed that serine phosphorylation of IRS1 was decreased at a target site of mTOR/p70 S6K, which has been associated with insulin resistance [158]. Recently, AMPK was found to phosphorylate the IR in response to glucose deprivation; conditions that also suppresses mTOR activity [146]. The α2 subunit of AMPK is required for AICAR-induced inhibitory effects of mTOR signaling in skeletal muscle [159], which is consistent with alternative findings that showing that under basal conditions the α2 subunit rather than α1 subunit is required for AICAR-induced glucose uptake [26]. Thus, increased AMPK activity could be hypothesized to impair inhibitory mTOR signaling towards IRS1. Further studies are required to establish this.
Adiponectin
The stimulation of AMPK by adiponectin may also be an alternative mechanism by which AMPK improves insulin sensitivity. Recent studies have shown that the activation of AMPK by adiponectin enhances the ability of insulin to stimulate IRS1 and Akt phosphorylation concurrently, with reduced S6K1 phosphorylation at T389 as well as IRS1 phosphorylation at S302 and S636/639 [160]. Aerobic training increases circulating adiponectin and adiponectin receptor mRNA in skeletal muscle [161], further supporting a role for AMPK signaling in improving insulin sensitivity during exercise. JNK and IKK can phosphorylate serine residues on IRS1 and impair insulin signaling; therefore, these studies may suggest that activation of AMPK during exercise may help restore insulin signaling that is impaired by these serine kinases in obesity.
IL-6
The gp130 receptor IL-6 protein was one of the first identified myokines whose production in skeletal muscle and release into the circulation increases almost 100 fold in response to exercise [162]. Circulating IL-6 levels are elevated in obesity and during exercise; however, in obesity high levels of IL-6 have been linked to insulin resistance whereas during exercise IL-6 may enhance glucose uptake via AMPK and improve insulin sensitivity given IL-6 is additive to insulin in promoting glucose uptake. Carey et al. [163] was one of the first studies to show that IL-6 increases glucose disposal in humans and glucose uptake in L6 myotubes; an effect dependent on AMPK signaling. Their study and Geiger et al. [164] also suggested that IL-6 is additive to insulin in stimulating glucose uptake; however, this was not observed by all studies [165] who found that IL-6 regulated glucose uptake and glycogen synthesis via a PI3-dependent pathway and not AMPK; given human myotubes targeted for AMPK depletion by siRNA did not have impaired insulin sensitivity. In contrast, Geiger et al. [164] showed that in whole muscle, which is more biologically relevant, that only higher concentrations of IL-6 increased AMPK phosphorylation; suggesting that under physiological conditions, such as during exercise, IL-6 may play a less significant role in positively regulating metabolic processes. The following year, Nieto-Vazquez et al. [166] found that the differential effects of IL-6 may be due to the amount of exposure to the cytokine, whereby, chronic treatment induces insulin resistance via mechanisms involving protein-tyrosine phosphatase 1B, JNK activation and SOCS3-mediated inhibition of insulin signaling, while acute exposure enhances insulin-stimulated glucose uptake via the LKB1/AMPK/TBC1D4 pathway. Recently, IL-6 KO mice reveal a relatively minor role for IL-6 in promoting glucose clearance during acute treadmill exercise [167]. Postexercise IL-6 levels are positively correlated with exercise duration (reviewed in [168]), and during the recovery period it could be speculated that the effects of IL-6 on metabolism are most pronounced. The majority of the data showing a positive effect of IL-6 on substrate utilization and insulin sensitivity has relied on the infusion of supraphysiological IL-6 concentrations, which does not demonstrate the physiological role of circulating IL-6 during and following exercise. Nonetheless finding from these studies have shown that IL-6 is important for insulin secretion and glucose tolerance via glucagon like peptide 1 (GLP-1) [169]. An alternative study utilizing IL-6 KO mice has shown that IL-6 is required for improving insulin-sensitivity in high-fat fed mice following chronic (4 weeks) voluntary wheel exercise training through lowering serum retinol binding protein 4 (RBP-4) levels [167], which have been implicated in obesity-induced insulin resistance in both humans and rodents [170,171]. Therefore, whilst these studies support a role for IL-6 in improving insulin sensitivity following exercise via RBP-4 and GLP-1, the involvement in AMPK in regulating this process remains to be determined.
CHRONIC EXERCISE TRAINING
Whilst it has been reported that AMPK activity and protein expression may be reduced in obesity [9,172,173], chronic exercise training increases AMPK protein expression and activity [174]. Therefore, AMPK may enhance insulin sensitivity through increasing TBC1D1 and TBC1D4 phosphorylation. Consistent with this idea, studies have also shown that chronic exercise training increases both protein expression and PAS phosphorylation of TBC1D4 in both humans and rats. TBC1D4 phosphorylation (S318, 341, 588, 642, and 751) is reduced in skeletal muscle from obese type 2 diabetics under basal conditions, but can be restored to comparable levels of nondiabetics by 10 weeks of endurance training [81]. Whether similar findings are observed for TBC1D1 is currently not known. Interestingly, a two week dietary intervention to induce weight loss improved insulin sensitivity in obese humans but did not alter TBC1D1 phosphorylation. Instead, the dietary intervention did increase TBC1D1 S231 and 660 but not S700 or PAS phosphorylation in response to exercise [175].
AMPK γ3 expression is reduced following strength training in obese type 2 diabetic humans and endurance training in healthy men [10,49]. Given deletion of the γ3 isoform is associated with impaired glycogen resynthesis following exercise; it is possible that reduced γ3 expression following exercise training is an adaptive response to enhance oxidative capacity and greater preference for lipid oxidation rather than glycogen utilization. It has recently been suggested that increased basal level of AMPK activity is likely due to γ1 associated complexes given γ3 expression is reduced [176].
CONCLUSIONS
In conclusion, AMPK plays an important role in regulating glucose uptake during exercise and may be involved in the insulin sensitizing effects of both acute and chronic exercise. A number of possible mechanisms have been proposed to play a role in insulin sensitizing effects of exercise; however, further studies are required to fully define the role of AMPK in mediating these effects. Exercise remains as one of the most cost effective treatments providing endless health benefits including both the prevention and treatment of obesity-induced insulin resistance. Therefore, future studies aimed at identifying the underlying mechanisms involved in the insulin sensitizing effects of exercise are essential for the development of new strategies both pharmacological and nonpharmacological such as exercise that target these diseases.


 XML Download
XML Download