

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
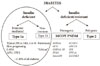
The current "etiological" classification of diabetes [1] is summarized in Fig. 1. Prior to the 1980s, nearly all children and young adults with diabetes were diagnosed with type 1 diabetes (T1D). Today, the proportions have changed, especially in Asian and African countries where more youths are diagnosed with type 2 diabetes (T2D) than T1D. Among the U.S. children 10 to 19 years old at diagnosis, half of African-American and Hispanic patients and more than half of Asian/Pacific Islanders and American Indians have T2D. However, the majority of diabetic non-Hispanic white adolescents still have T1D and nearly all children with diabetes diagnosed under age ten have T1D [2].
With rising obesity rates in children, it is increasingly difficult to differentiate between T1D and T2D on clinical grounds alone. Islet autoantibodies, fasting or stimulated C-peptide levels, and genetic markers provide tools to augment a clinical diagnosis. Globally, the incidence of T1D is increasing by up to 5% per year [3,4], doubling approximately every 20 years [5,6]. Over the past 20 years, newly industrialized countries have experienced an epidemic of T1D, mirroring that of T2D, but likely caused by entirely different environmental agent(s).
PATHOMECHANISMS OF TYPE 1a (AUTOIMMUNE) DIABETES (T1aD)
Diabetes is a heterogeneous group of diseases with the common feature of hyperglycemia, however, resulting from combinations of defects in at least 40 genes and a variety of environmental agents. T1aD is caused by lack of insulin due to autoimmune destruction of the pancreatic islet beta-cells. The immune system fails to maintain tolerance to beta-cell autoantigens, often in the setting of the HLA-DRB1*03, DQB1*0201, DRB1*04, DQB1*0302, and/or the HLA-DRB1*0901, DQB1*0303 haplotypes. Chronic inflammation in the islets leads, usually after years, and rarely in just months or days, to insulin-dependent diabetes. T1aD is defined by the presence of autoantibodies to the beta-cell antigens detected before, at or after clinical diagnosis: autoantibodies to insulin (IAA), the tyrosine phosphatase insulinoma antigen (IA-2A), glutamic acid decarboxylase (GADA), and zinc transporter 8 (ZnT8A). One and usually more of these autoantibodies are present in 85% to 95% of newly diagnosed T1aD patients, but this proportion varies depending on patient's age, the number and quality of the assays used, and ethnicity. A small number of T1aD patients may be negative for all islet autoantibodies at diagnosis, despite presence of the autoantibodies prior or after diagnosis (our own observations from the Diabetes Autoimmunity Study in the Young [7]).
The rate of β-cell destruction is quite variable - rapid in younger children and those with high risk HLA genotypes, especially DRB1*03, DQB1*0201, DRB1*04, DQB1*0302, and slower in adolescents and adults and those with lower-risk HLA genotypes. This may explain a higher proportion of slowly progressing T1D reported in Asian and African populations where a larger proportion of the patients carry neutral or protective HLA-DR, DQ genotypes. In Japan, ~10% of children with diabetes are diagnosed with "slowly progressing T1D." Most are picked on a school-children screening for glucosuria. While their progression to full insulin dependence is slower, 90% have islet autoantibodies or HLA genotypes consistent with classical T1aD (Nan Tajima, personal communication, 2010).
Some patients, particularly children and adolescents, may present with ketoacidosis as the first manifestation of the disease. Others have modest hyperglycemia that can rapidly decompensate in the presence of infection or other stress. Still others, particularly adults, may retain residual β-cell function sufficient to prevent ketoacidosis for many years; such individuals eventually also become dependent on insulin for survival and are at risk for ketoacidosis. At this latter stage of the disease, there is little or no insulin secretion, as manifested by low or undetectable levels of plasma C-peptide. Immune-mediated diabetes commonly occurs in childhood and adolescence, but it can occur at any age, even in the 8th and 9th decades of life. In Western countries, more than half of T1D patients are diagnosed after the age of 20 years.
MONOGENIC AUTOIMMUNE POLYENDOCRINE SYNDROMES (APS)
APS1 is a rare syndrome caused by a truncation of the autoimmune regulator gene (AIRE). The syndrome is defined by the combination of Addison's disease, hypoparathyroidism, and mucocutanous candidiasis; oophoritis is quite common. T1aD is present in ~20% of the cases.
Immunodeficiency, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome is caused by one of several possible mutations in the FOXP3 gene [8] on the X chromosome. Only males are affected. The patients develop chronic enteropathy, eczema, autoimmune thyroid disease, anemia, and T1aD (in about 60% of the cases).
PATHOMECHANISMS OF TYPE 1b (IDIOPATHIC) DIABETES (T1bD)
Some forms of type 1 diabetes demonstrate permanent insulinopenia and proneness to ketoacidosis, but no evidence of autoimmunity. Only a small proportion of T1D patients fall into this category, of those who do, most are of African or Asian ancestry.
FULMINANT T1bD
Fulminant type 1 diabetes was first described by Japanese investigators in 2000 [9] and later reported in Korean [10-13], Chinese [14], French [15], and the U.S. Hispanic patients [16]. Typical features of this syndrome include: severe hyperglycemia and DKA shortly after the onset of diabetic symptoms and normal HbA1c levels. A rapid and almost complete destruction of beta cells leads to low C-peptide levels, no remission period, and a complete insulin dependency. High frequency of flu-like symptoms has been reported as well as elevated pancreatic enzymes, pointing perhaps to a primary infection of the exocrine pancreatic tissue. Pregnant women appear to be disproportionally affected. While fulminant diabetes is believed to be distinct from typical T1aD, insofar that chronic islet autoimmunity contributes less critically to the beta-cell damage than a hypothesized viral infection, there appears to be a significant overlap between these two types of diabetes in their HLA-DR, DQ associations and the presence of insulitis with beta-cell antigen-reactive T-cells. Interestingly HLA-identical dizygotic twins have been reported, one with typical T1aD and the other with fulminant diabetes [17] diagnosed at different times. While GADA was reportedly "infrequent" (<5%) and IA-2A absent, none of the studies so far have measured IAA or ZnT8 levels. It appears that this syndrome perhaps represents an extreme end of the T1aD spectrum, rather than an etiologically distinct disease. The syndrome remains rare, affecting <1% of children with diabetes in Japan (Nana Tajima, personal communication, 2010).
KETOSIS-PRONE T1bD
Several reports from sub-Saharan Africa [18-21] and the U.S. African-American population [22-25] have described atypical ketosis-prone "African" diabetes. While this clinical entity remains poorly defined, the common characteristics appear to include acute onset, often with ketoacidosis, in often obese adolescents or young adults with no islet autoantibodies and HLA genotypes inconsistent with T1aD. The post-diagnosis course is characterized by nearly complete remissions followed by slow progression to insulin dependence with periods of normoglycemia intertwined with episodes of hyperglycemia and ketosis requiring insulin. Strong family history of diabetes and male predominance has been reported, while age at onset and body mass index (BMI) have varied across studies. Preliminary data from nPOD [26] showed no pseudoatrophic islets characteristic to T1aD, confirming clinical observations of C-peptide levels preserved many years after diagnosis. While some believe that in Africa ketosis prone T1bD is more frequent than the classical T1aD, more of untreated T1aD than T1bD patients may be dying prior to or shortly after diagnosis.
CHALLENGES IN THE INTERPRETATION OF TESTS USED TO DIAGNOSE T1D
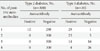
The clinical and laboratory characteristics used currently to distinguish T1D and especially T1aD are summarized in Table 1. Their advantages and limitations are reviewed below.
Islet autoantibodies
Autoantibodies to biochemically characterized beta-cell autoantigens: IAA, IA-2A, GADA, and ZnT8A [27] help to define T1a, if measured prior to or shortly after initiation of insulin therapy. IAA are masked by antibodies induced by exogenous insulin and become very hard to measure after just 10 to 14 days of insulin therapy. ZnT8A tend to disappear quickly after diagnosis of diabetes, while GADA and IA-2A tend to persist longer, but are rarely seen more than 5 years after diagnosis. Testing for at least two of these autoantibodies at diagnosis is now considered standard of care in T1D. Good commercial assays exist for IA-2A, GADA, and ZnT8A, with the former two recently harmonized [28]. IAA are low-affinity antibodies and harder to measure; however, high-quality non-radioactive assays for IAA are close to being commercially available [29]. The search for additional islet autoantibodies and assay that would reliably detect autoreactive T-lymphocytes are active areas of research.
The prevalence of antibodies in patients with T1D varies depending on the study population and methods of antibody assessment. In the multi-center SEARCH trial of newly diagnosed cases of T1D and T2D in youth 0- to 19-year-old, 52% were positive for GADA, 60% were positive for IA-2A and 38% were positive for both [30]. This study did not test for IAA or ZnT8A and obtained the blood sample months to years after diagnosis. In contrast, the Childhood Diabetes in Finland Study Group found that among newly diagnosed children with T1D, 91% tested positive for at least two antibodies and 71% for three or more. Further, IA-2A was detected in 86% of cases [31].
We recommend that as many biochemical islet autoantibody assays as possible (up to 4) are used while confirming the diagnosis of T1aD. If only GADA and IA-2A are measured - up to 20% of truly T1aD patients will be missed as they have only IAA or ZnT8A; the latter should be measured immediately at diagnosis. Positivity for one of the autoantibodies usually suffice; positivity for 2 or more removes any doubt concerning diagnosis of T1aD (see below).
Islet autoantibodies can by falsely positive as often as patients with T2D as in non-diabetic persons. Let us consider that a clinician is using one assay, e.g., GADA, that is 95% specific for T1aD. In a mixed patient population of 300 T2D subjects and 30 T1aD patients, 35% of positive GADA results will be false-positive. However, using a better assay, with a 99% specificity, would result in only 10% of false-positive results.
The likelihood of obtaining false-positive results increases with application of multiple assays. Table 2 illustrates results of testing of the same mixed population of patients with T2D and T1aD using four islet autoantibody assays, each characterized by 60% sensitivity and 99% specificity. Bayesian probability of a false-positive finding in T2D patients is 12/300 (4%), however, none of the T2D patients would be expected to be positive for more than one autoantibody. The same probabilities of false-positive findings would apply to 300 non-diabetic subjects screened for islet autoantibodies. Please note that even with four excellent assays, one (3.3%) of the 30 T1aD patients is expected to be negative for all autoantibodies and only 4 would be positive for all four.
C-peptide levels
Nearly half of adults with T1aD have significant C-peptide levels within 5 years of diagnosis, and 8% had significant C-peptide 5 to 15 years after diagnosis [34]. The rate of C-peptide disappearance in T1aD patients diagnosed in their 30's is ~20% to 30% per year, slower than that among teenagers (45% to 55% per year) or younger children (60% to 90% per year). Thus persistence of beta-cell function is rare in young children with T1aD, especially those with the highest-risk HLA-DR, DQ genotypes and multiple islet autoantibodies. While measurement of C-peptide levels post stimulation with mixed meal or intravenous glucagon remains the gold standard for clinical trials, fasting C-peptide levels are more readily available in clinical practice for differential diagnosis of T1D vs. T2D. The 5th and 50th percentiles of fasting C-peptide in healthy adolescents, aged 12 to 19 years, who participated in the National Health and Nutrition Survey 1999 to 2002 were, respectively, 1.0 and 1.9 ng/mL [35] while the levels ≥0.23 ng/mL are considered clinically helpful in lowering the risk of long-term complications (J. Lachin, P.F. McGee, DCCT, unpublished data). The SEARCH study has demonstrated [36] that the current classification defining T1D as a state of absolute insulin deficiency and T2D as a state of insulin resistance combined with inadequate insulin secretion (Fig. 1) is inadequate. During the 1st year after diagnosis, almost one-third of children with T1aD had C-peptide values that exceeded the 5th percentile and 7% exceeded the 50th percentile for healthy adolescents [36]. Thus, health care providers should be careful not to use normal C-peptide levels as a guide to delay insulin treatment in patients with the T1aD phenotype. Practically speaking, while absent or low C-peptide confirms diagnosis of T1D in islet autoantibody negative patients, normal C-peptide levels are expected in either T1D or T2D patients.
Genetic markers
Individuals with the HLA-DRB1*03, DQB1*0201/DRB1*04,DQB1*0302 genotype are at approximately 20-fold increased risk for T1aD compared to the general population. This high risk genotype is present in 2.4% of newborns of European ancestry [37], but less frequent in Africans or Asians [38]. By age 15, 5% of children with this genotype will develop islet autoimmunity and T1D, compared with only 0.3% in the general population. A number of additional HLA class II genotypes confer moderately increased risk for T1D, while others are protective.
Non-HLA associated loci that result in increased risk of T1D include those that influence immunity (INS, PTPN22, IL2RA, SH2B3), insulin production and metabolism (ERBB3), and many others. A number of novel loci identified through genome-wide association studies have been confirmed in prospective population-based studies. However, jointly they confer only a small additional risk compared to the effect of HLA-DR and DQ. An up-to-date review of all genes implicated in the development of T1D can be found at T1Dbase (http://www.t1dbase.org).
In practice, genotyping is rarely used to distinguish T1aD from other forms of diabetes. However, HLA-DR, DQ typing is worth of consideration in autoantibody negative cases where other factors, e.g., clinical course, family history or presence of other autoimmune diseases suggest T1aD. Among islet autoantibody negative diabetic patients younger than 20, at the Barbara Davis Center in Colorado, 32% of non-Hispanic whites had HLA-DR, DQ genotypes compatible with T1aD, compared to only 13% Hispanic, and 5% African American patients. More work is needed to fully utilize genetic markers, especially in non-European ethnic groups. Non-HLA class II markers are likely of little value, except for highly specific AIRE or FOXP3 mutations, if clinical picture suggests the APS1 or IPEX, respectively.
Family history of T1aD
At the time of diagnosis, over 85% to 95% of patients with T1D lack a family history of the disease in immediate relatives. This proportion decreases over time, as by the time the patients reaches age 40, in about 10% of the families a sibling, parent or offspring develops T1D [39,40]. Thus, while T1D among first-degree relatives of a patient strongly suggest diagnosis of T1D, negative family history does not help to rule it out.
Obesity
The presence of obesity is compatible with diagnosis of T1aD. In fact, about half of the patients with BMI ≥27 diagnosed with diabetes before the age of 20 in the U.S. had T1aD (Fig. 2).
Insulin sensitivity
Several lines of evidence support the hypothesis that insulin resistance may accelerate progression to over hypoglycemia among persons with islet autoimmunity and significant beta-cell defect, however, the independent effect of insulin resistance on progression to T1aD appeared to be modest [41]. The SEARCH study in the U.S. attempted to classify cases of childhood diabetes using the presence of GADA and/or IA-2A as well as insulin sensitivity estimated from the patient's waist circumference, HbA1c and triglyceride levels [30]. Not unexpectedly, children who were positive for islet autoantibodies were similar genetically, in terms of their C-peptide levels, presence of DKA or clinical course post diagnosis regardless of being classified as "insulin resistant" or "insulin sensitive." Insulin sensitivity is a fleeting phenotype that may changes within weeks, even days, with changes in physical activity, diet composition, body weight, and hyperglycemia levels. Most T1aD patients are insulin resistant compared to BMI-age-matched non-diabetic controls [42]. In addition, indices of insulin sensitivity derived from clinical variables correlate poorly with insulin sensitivity levels measured directly using euglycemic hyperinsulinemic clamp. Therefore, assessment of insulin sensitivity, even using invasive methods, is unlikely to be of help in ruling out T1D.
Presence of other autoimmune diseases
Due to shared genetic susceptibility, T1aD often coexists with autoimmune disorders such as celiac disease, Hashimoto's thyroiditis, Graves' disease Addison's disease, vitiligo, autoimmune hepatitis, myasthenia gravis, and pernicious anemia. Presence of any of these conditions or serologic evidence of subclinical autoimmunity, e.g., autoantibodies against tissue transglutaminase, thyroid peroxidase, 21-hydroxylase or parietal cells, may aid correct classification in borderline cases.
CONCLUSIONS
With increasing rates of obesity, it is becoming increasingly difficult to distinguish between T1D and T2D. In absence of islet autoantibodies, it may take weeks or months of observation to accurately diagnose a child with features of both T1D and T2D. It must be noted that for a child presenting with sustained hyperglycemia or DKA, insulin therapy must precede definitive laboratory results, even in ambiguous cases. Fig. 3 summarizes our experience in classifying diabetes into T1a, T1b, and T2 in the multiethnic population of patients diagnosed below age 20, in Colorado, the USA.
The scheme is based primarily on islet autoantibody testing, with the aid of HLA-DR, DQ genotyping and fasting C-peptide measurement in autoantibody negative subjects. While the genetic markers used will need to be customized for other ethnic groups, especially of Asian or African origin, this scheme is likely to minimize expense and maximize accuracy of assigning the correct type of diabetes in different populations.



 XML Download
XML Download