

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Diabetes mellitus (DM) is a scourge to the global community, stepping up at a magnanimous proportion. As per the data provided by International Diabetes Federation, the worldwide prevalence of DM in 2011 was 366 million and this number is projected to reach 552 million by 2030 [1]. Type 2 diabetes mellitus (T2DM) follows as a result of impaired glucose homeostasis. Insulin resistance (IR) is measured by glucose homeostasis model assessment (HOMA), first described by Matthews and colleagues [2]. T2DM is characterized as a non-autoimmune condition, which involves multiple, intriguing factors like genetics, environmental or acquired factors and presence of inflammatory pathways. IR is a complicated condition involving multiple etiological pathways. It plays a crucial part in the pathogenesis of metabolic syndrome and T2DM, yet the inherent mechanisms are not completely cognizant [3]. Periodontitis is the most common oral infection with wide global prevalence. Clinical features of periodontitis include bleeding gingiva, increased interdental spacing, increase in probing depth, bad oral breath and mobility of teeth in advanced cases. Since periodontitis is asymptomatic, the affected subjects are largely unaware and refrain from periodontal treatment. Periodontitis is characterized by the loss of tooth supporting tissues, which is indolent in nature with marked chronicity. The primary etiology of periodontitis is dental plaque, which houses multiple bacteria of different strains and species [4]. It has been proven that periodontitis has effects, impacting the systemic health of the subject and the detrimental effects are not only confined to the oral cavity [5]. Periodontal medicine is an emerging branch which addresses the various links of periodontitis with systemic diseases. Periodontitis DM maintain a "two way relationship" [6]. The present review addresses the issue of IR in particular and the potential causal role of periodontitis in pathogenesis of the same.
ETIOPATHOGENESIS OF PERIODONTITIS AND ITS SYSTEMIC LINK
Periodontitis is essentially a biofilm induced disease, initiated and progressed by different bacterial species, present in the dental plaque. The periodontopathic bacteria are basically gram-negative in nature and they are present in the depths of periodontal pockets, placed at low oxygen tension. The putative pathogenic bacteria express noxious toxins instrumental for the periodontal destruction [7]. Currently, the consensus regarding pathogenesis of periodontitis has undergone an immense change. According to this concept, periodontitis is not only the result of adverse microbial activity but as an interaction among various other factors like genetics, systemic health, immunity, environmental factors like tobacco and stress. The above mentioned factors play an important role in the modification of host response to the disease process. Thus, sometimes the periodontal disease may exhibit varied expression [7]. Various pro-inflammatory mediators like interleukin (IL)-1α and IL-1β, IL-6, tumor necrosis factor (TNF)-α, prostaglandin E2 (PGE2), matrix metalloproteinases are expressed in periodontitis, as a result of activation of the host immune-inflammatory mechanisms. Cytokines are liberated by periodontal tissues like fibroblasts, endothelial cells, macrophages, osteoclasts, epithelial cells, neutrophils, monocytes, lymphocytes, and mast cells. Immune cells like neutrophils, monocytes also let out cytokines in inflammatory conditions. This host tissue expressed array of factors may be detrimental to the host tissue itself, amplifying the destructive disease process [8]. The periodontopathogenic flora produce toxins and significant challenge is offered by lipopolysaccharide (LPS), a component of the gram-negative bacterial cell wall. LPS is a potent endotoxin which exacerbates the host inflammatory response. Subjects with periodontitis are reported to present endotoxin activity in the serum [9]. As discussed previously the bacteria are amicably housed in the periodontal pocket. These bacteria, attended with their noxious products can gain a ready access through the ulcerated lining of the periodontal pocket, into the systemic circulation. Loos [10] reported a significant cumulative surface area of all periodontal lesions in a patient with severe periodontitis, ranging from 15 to 20 cm2. Further, the periodontal inflamed surface area (PISA) can be used as a tool to accurately assess the amount of periodontal inflamed tissue in a subject with periodontitis [11]. Thus, it can be inferred that, in severe periodontitis patients, pro-inflammatory mediators (IL-1α and IL-1β, TNF-α, PGE2) from the disease gingival sites may be 'poured' into the systemic circulation. Studies have identified many systemic biomarkers, exposing the link of periodontitis with systemic conditions and cardiovascular disease [12,13]. Thus, it can be enunciated that periodontitis is a "low grade infection" capable of developing a "low grade systemic inflammation" with an ability to influence the general systemic health.
ETIOPATHOGENESIS OF IR AND POTENTIAL LINK WITH PERIODONTITIS
IR, a precursor to T2DM has a complicated metabolic mechanism, with multiple etiological pathways. It is proposed that a defect in insulin receptor substrate (IRS) protein function is necessary for the uncoupling of the insulin signal, resulting in IR [14]. Various protein kinases, which are important in insulin signaling, are key players in IR [15]. Insulin functions by binding to the heterotetrameric membrane receptor leading to IRS-1 phosphorylation and IRS-1-associated phosphatidylinositol 3 phosphate kinase (PI3 kinase) activation [16]. This event in turn impacts effectors like Akt/protein kinase B (PKB), which triggers the glucose transporter GLUT4. GLUT4 is further translocated into the membrane and induces glucose import into the cell [17]. Protein kinase C (PKC) isoenzymes is a family of signaling molecules involved in the actions of insulin. These PKCs are categorized as classical isoenzymes, novel isoenzymes, and atypical isoenzymes [18]. They operate in an elaborate manner and are noted to play a positive and negative regulatory role in insulin signaling [19,20]. Signal activity from a stimulus to the ordinance of cellular processes, considering those involved in glucose homeostsis, mainly depends upon protein kinase signaling. A defect in the insulin signaling process results in downregulation of Akt/PKB, thereby inhibiting the required cascade process. Kinases like Jun N-terminal kinases (JNKs), also named SAPKs can phosphorylate IRS-1 & 2 at specific serine and threonine residues, leading to suppression of insulin signaling [21]. Peroxisome proliferator-activated receptor (PPAR)-γ complements insulin signaling. Pro-inflammatory cytokines increase the IR, by activation of JNK and IκB kinase-β/nuclear factor κB and downregulation of PPAR-γ [22,23].
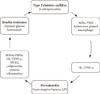
A "bidirectional relationship" between periodontitis and DM has been proposed (Fig. 1). It is theorized that pro-inflammatory cytokines expressed by gingiva in periodontitis enter the systemic circulation leading to exacerbation of DM. Conversely, the elevated levels of the pro-inflammatory cytokines in DM may reach the gingiva leading to aggravation of already existing periodontal disease [24,25]. Chronic subclinical systemic inflammation may be conducive to impaired glucose homeostasis/increased IR, subsequently paving a way for clinical manifestation of T2DM [23].
Chronic hyperglycemia in T2DM, indulges nonenzymatic glycation of proteins with the formation of advanced glycation end products (AGEs), which are reported to prime the macrophages to express cytokines (IL-6 and TNF-α). These cytokines are instrumental in the release of acute phase reactants (CRP) from the liver, further amplifying the existing inflammation [26]. Currently, adipose tissue is regarded as an endocrine organ, a major depot, capable of secreting bioactive agents called adipokines [27]. Adipokines enlisted in regulation of IR are adiponectin, leptin, resistin, visfatin, chemerin, TNF-α, IL-1, IL-6, IL-8, IL-10, plasminogen-aktivator-inhibitor-1, monocyte chemoattractant protein-1, and retinol binding protein-4 [28]. Adiponectin maintains a mutual antagonistic action to TNF-α, plays an important role as anti-diabetic, anti-atherogenic, anti-inflammatory agent. Studies report that TNF-α inhibits the expression of adiponectin. Conversely, adiponectin suppresses LPS induced TNF-α production [29,30]. TNF-α is one of the most important cytokine implicated in the initiation and progression of IR.
Table 1 depicts the possible mechanisms by which TNF-α contributes to IR [31-38]. The role of IL-6 in IR is controversial as reported in the literature [39,40]. It can be inferred that relentless increase in the systemic levels of IL-6, as in obesity and T2DM may lead to IR, whereas a transient increase in IL-6 may assist in normal glucose homeostasis [41]. With the new entropy in the advancement of molecular biology an increased onus is laid upon the role of inflammatory mediators in the pathogenesis of IR and subsequent T2DM. Periodontitis and T2DM share a common process of pathogenesis, involving inflammatory response at the local and systemic level [42]. Studies directed at assessing the IR utilize HOMA for estimation of insulin sensitivity. The HOMA method deduces an estimate of insulin sensitivity from a mathematical model of fasting plasma glucose and insulin concentrations [2].
Table 2 outlines the studies relating IR with periodontitis. The studies depicted in the table are animal and human studies [43-49]. Obesity has also been portrayed as a condition associated with low grade systemic inflammation, demonstrating commonality in the expression of identical cytokines as that detected in periodontitis and T2DM [46]. T2DM subjects with concomitant periodontitis exhibit increased biomarkers and oxidative stress. Compromised β-cell function and increased IR is attributed to the intensified oxidative stress as a result of hyperactivated neutrophils in periodontitis, ensuing in the boosted release of reactive oxygen species [49,50].
A common denominator that exists for periodontitis and T2DM is the systemic presence of common pro-inflammatory cytokines. Obesity is now believed to bear a causal relationship with periodontitis [51,52]. A bidirectional association between DM and periodontitis has been emphasized by abundant literature [6,53,54]. It is proposed that periodontitis and IR are potential risk factors for the perturbation of cardiovascular health [55-57]. IR is an important component of metabolic syndrome and it is proposed that periodontal disease should be considered as a component of metabolic syndrome [42,58]. It is important to acknowledge periodontitis, as an emerging risk factor for metabolic syndrome. There is still a sizeable vacuum in evidence based literature, with regards to the connection of periodontitis and IR. This issue has not been adequately addressed in majority of the studies. The studies in rodent models are worthy for realizing the potential cellular mechanisms of the pathogenesis with IR, but there is still doubt whether the pathways and trials in these animal models can be extrapolated in humans. The PISA contributes as a dynamic source for the progression of poor metabolic control in T2DM subjects with severe periodontitis. Nesse et al. [59] have calculated that an increase of PISA with 333 mm2 was associated with a 1.0 percentage point augmentation of HbA1c, independent of other factors. A similar study should be conducted which can indicate a dose-response relationship between PISA and IR. PISA can serve as a valuable tool to quantify the inflammatory burden in periodontitis and relate it with IR.
CONCLUSIONS
Pro-inflammatory cytokines amplify IR. IR may be a constituent of the causal pathway connecting inflammatory mediators to incident diabetes. There is a lack of research in human subjects concerning periodontitis, as a causal pathology for IR. Periodontitis and IR are largely unrecognized. Hence it is connoted, to perceive the early presence of IR and periodontitis. Both the conditions existing conjointly in the same individual, can reciprocate the pernicious effects of each other. Extensive, multicentric, randomized controlled trials involving large populations are vindicated to analyze the elusive link between periodontitis and IR. The potential effects of periodontal therapy in the de-escalation of IR should be contemplated. The potential favorable benefits of anti-cytokine therapy to treat IR should be explored. Although, the high prevalence of periodontitis in diabetics is cognizant by the dental professionals, it is not a well known fact in the medical community. Periodontitis should receive due attention as a "pandemic" by the respective national and world health governance. Both, periodontitis and DM, are cryptically linked to metabolic syndrome and cardiovascular diseases. The medical and oral health professional should align efforts in management of T2DM susceptible subjects with periodontitis. Concerted endeavors can be valuable to control the progression of both the conditions respectively.


 XML Download
XML Download