

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The prevalence of obesity continues to increase at an alarming rate around the globe. The World Health Organization has forecasted that approximately 2.3 billion adults worldwide will be overweight and more than 700 million will be obese by 2015 [1]. Since obesity is associated with increased risks for type 2 diabetes, cardiovascular events, stroke, certain types of cancer, and neurodegenerative diseases [2], an obesity epidemic will threaten human health in the upcoming years.
Obesity is a state in which energy intake exceeds energy expenditure over a prolonged period. Food intake is promoted by hormones signaling hunger, the availability of high calorie palatable foods, and learned food preferences. It is inhibited by leptin and other hormones that generate satiety, including insulin and gut-derived hormones. A chronic imbalance between hunger and satiety signals leads to long term alterations in food intake and body weight.
BRAIN AREAS INVOLVED IN FEEDING REGULATION
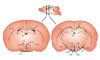
Hypothalamus
The hypothalamus, a small area of the brain located just below the thalamus, is the regulating center of appetite and energy homeostasis. The hypothalamus consists of several interconnecting nuclei: the arcuate nucleus (ARC), paraventricular nucleus (PVN), lateral hypothalamic area (LHA), ventromedial nucleus (VMN), and the dorsomedial nucleus (DMN) (Fig. 1). The ARC of the hypothalamus is adjacent to the median eminence, a circumventricular organ having defective blood-brain barriers (BBB). Thus, circulating hormones and nutrients can access the ARC without passing the BBB. Moreover, the ARC surrounds the third cerebroventricle. Hormones and nutrients in the cerebrospinal fluid can diffuse into the extracellular fluids of the ARC. Due to these anatomical features, the ARC is considered to be a hypothalamic area primarily sensing peripheral metabolic signals. In the ARC, there are two distinct neuronal populations: one is a group of neurons coexpressing orexigenic neuropeptides, including neuropeptide Y (NPY) and agouti-related peptide (AgRP), and the other is a subset of neurons expressing anorexigenic neuropeptides, including proopiomelanocortin (POMC) and cocaine- and amphetamine-regulated transcript (CART). These neurons are first-order neurons where peripheral metabolic signals including leptin, insulin, ghrelin, and nutrients are primarily transferred. Anorexigenic monoamine serotonin also acts on POMC neurons through the 5HT-2C receptor to induce anorexia [3]. POMC neurons send axonal projections to the second-order neurons in other hypothalamic areas, the PVN, VMN, and LHA.
The α-melanocyte-stimulating hormone (α-MSH), an anorexigenic neuropeptide, is produced by the posttranscriptional processing of POMC and released from presynaptic terminals of POMC neurons. By binding to the melanocortin-3 and -4 receptor (MC3R, MC4R) on the second order neurons, α-MSH activates catabolic pathways: reduced food intake and enhanced energy expenditure [4]. Targeted deletion of the MC4R in mice resulted in hyperphagia, reduced energy expenditure, and obesity [4]. In humans, MC4R mutations account for about 6% of severe early-onset obesity [5], supporting an important role for the central melanocortin system in the control of energy metabolism.
Endogenous melanocotin receptor antagonist AgRP is released from the terminals of ARC NPY/AgRP-producing neurons to the synaptic space on the second order neurons where it competes with α-MSH on MC3R and MC4R and antagonizes the effects of α-MSH [6]. Selective ablation of NPY/AgRP neurons in young mice resulted in a significant decrease in food intake and body weight [7], suggesting that these neurons are critical for promoting food intake and preventing weight loss.
The PVN neurons synthesize and secrete neuropeptides that have a net catabolic action, including the corticotrophin-releasing hormone, thyrotropin-releasing hormone, somatostatin, vasopressin, and oxytocin. In addition, PVN sends sympathetic outflow to the peripheral metabolic organs, including liver and adipose tissue, resulting in increased fatty acid oxidation and lipolysis [8]. Destruction of PVN and haploinsufficiency of Sim1, a critical transcriptional factor in the development of PVN, caused hyperphagia and obesity [9], implying a inhibitory role for PVN in food intake and weight gain.
The VMN mainly receives neuronal projections from the ARC, and projects their axons to the ARC, DMN and LHA, as well as brainstem regions. The VMN contains neurons that sense glucose and leptin [10]. Moreover, anorexigenic neuropeptide, a brain-derived neurotrophic factor (BDNF), is produced in the VMN [11]. Destruction of the VMN caused hyperphagia and obesity, as well as hyperglycemia [12]. Thus, the VMN is regarded as a pivotal area in generating satiety and maintaining glucose homeostasis. The DMN contains a high level of NPY terminals and α-MSH terminals originating from the ARC [13]. Destruction of DMN also results in hyperphagia and obesity [14].
In contrast to PVN, VMN, and DMN, destruction of LHA leads to hypophagia and weight loss. Therefore, LHA has been considered to be a feeding center. LHA contains two neuronal populations producing orexigenic neuropeptides, the melanin concentrating hormone (MCH) and orexin, also called hypocretin. NPY/AgRP- and α-MSH-immunoreactive terminals from ARC neurons are in contact with MCH- and orexin-expressing neurons. Orexin-producing neurons are also involved in glucose sensing and the regulation of sleep-awake cycles [15]. Mice with orexin receptor 2 displayed canine narcolepsy. On the other hand, depletion of MCH or the MCH 1 receptor in mice attenuated body weight, suggesting that MCH acts as endogenous orexigenic molecules [16].
Brainstem
The brainstem is another key brain area involved in regulation of food intake and energy balance. Satiety signals from the gastrointestinal (GI) tract primarily relay to the solitary tract nucleus (NTS) through the sensory vagus nerve, a major neuronal link between the gut and the brain. Transaction of sensory vagal fibers resulted in increased meal size and meal duration, confirming that vagal afferents transfer satiety signals to the brain [17]. Like the ARC, the NTS is anatomically close to the circumventricular organ area postrema (AP). Therefore, the NTS is located in a perfect place for receiving both humoral and neural signals. Meanwhile, the NTS receives extensive neuronal projections from the PVN and vice versa, indicating that there is intimate communication between the hypothalamus and the brainstem. Similarly to hypothalamic neurons, NTS neurons produce glucagon-like peptide 1 (GLP-1), NPY, and POMC, as well as sensing peripheral metabolic signals. For instance, POMC neurons in the NTS show the signal transduction activated transcript 3 (STAT3) activation in response to leptin [18]. Thus, circulating hormones and nutrients may inform metabolic signals to the brain by acting on the hypothalamus and brainstem.
Midbrain
The brain rewarding system is involved in the control of hedonic feeding, i.e., the intake of palatable foods. Like other addition behaviors, the mesolimbic and mesocortical dopaminergic pathways are involved in hedonic feeding. Intake of palatable foods elicits a dopamine release in the ventral tegmental area (VTA), which in turn activates the neural pathways from the VTA to the nucleus accumbens (NA) via the medial forebrain bundles. Interestingly, hedonic feeding is modulated by metabolic signals. Leptin acts on the dopaminergic neurons in the VTA to suppress feeding [19]. Conversely, hedonic feeding can override satiety signals. Mice lacking a D2 receptor were more sensitive to leptin [20].
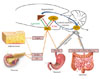
PERIPHERAL ADIPOSITY SIGNALS
Leptin
The obese gene coding leptin was first identified by positional gene cloning of ob/ob mice in 1994 [21]. Leptin is exclusively produced in white adipocytes and released to systemic circulation. Plasma leptin concentrations increase in proportion to body fat mass and thus can be used as biomarker of adiposity. Circulating leptin enters the brain through BBB and the blood-CSF barriers through receptor-mediated mechanisms. Leptin receptors are highly expressed in the neurons of the hypothalamus, especially the ARC. Leptin binds the long form leptin receptors, Ob-Rb, on the ARC neurons which subsequently induces activation of Janus kinase 2 (JAK2)-STAT3 signaling and inhibition of AMP-activated protein kinase (AMPK) activity [22]. Activation of hypothalamic leptin signaling causes an increase in neuronal activity of POMC/CART neurons while it decreases activity of NPY/AgRP neurons [23], resulting in reduced food intake and enhanced energy expenditure. Interestingly, leptin is also produced in the gastric epithelium and locally amplifies gut satiation signals such as cholecystokinin (CCK) [24]. Leptin also affects the thresholds of sweet taste perception in the tongue [25].
Leptin administration has successfully treated hyperphagia and obesity in humans and rodents with leptin deficiency [26]. However, most obese humans have elevated plasma leptin levels, implying they may have leptin resistance rather than leptin deficiency. Moreover, leptin treatment in obese subjects has proven to be ineffective. One possible mechanism underlying leptin resistance is reduced leptin transport to the brain, which may be due to saturation of leptin transporters at the BBB [24]. Furthermore, elevated plasma proinflammatory cytokines and free fatty acids in obese subjects may impair leptin transport [27]. On the other hand, leptin resistance may result from reduced leptin signaling in hypothalamic neurons. Notably, leptin-induced STAT3 activation was selectively impaired in the hypothalamic ARC [28]. Several mechanisms, including the suppressor of cytokine signaling (SOCS)-3, protein tyrosine phosphatase (PTP)-1B, I-kappa B kinase (IKK), nuclear factor-kappa B (NF-κB), c-Jun kinase (JNK), endoplasmic reticulum stress, and defective autophagy have been shown to contribute to impaired leptin signaling in the hypothalamus of obese mice [29].
Insulin
Insulin is rapidly secreted from pancreatic β-cells following a meal and transported to the brain. Fasting plasma insulin levels have a good positive relation with body fat mass. Thus, insulin is considered to be a surrogate marker for adiposity. In the CNS, insulin receptors are expressed in hypothalamic nuclei, such as the ARC, DMN, and the PVN, well-known areas involved in feeding regulation [30]. Like leptin, insulin binds insulin receptors on ARC neurons, resulting in activation of POMC neurons and inhibition of NPY/AgRP neurons through the insulin receptor substrate (IRS)-2, the phosphatidyl inositol-3-kinase (PI3K)-Akt-FoxO1 signaling pathway [31]. Through these effects, insulin relays an anorexigenic signal to the brain. The role of insulin in the regulation of energy balance was supported by finding that deletion of the neuron-specific insulin receptor and IRS-2 causes an obesity phenotype in mice [32].
APPETITE REGULATING GI HORMONES
The GI tract is considered to be the largest endocrine organ in the body. In addition to its original function as a digestive and absorptive organ, the gut plays an important role in the control of energy homeostasis, particularly in short-term regulation of food intake.
Cholecystokinin (CCK)
CCK is the first gut hormone which has been shown to have anorexigenic action [33]. Intravenous injection of CCK reduces meal size and duration in humans and rats [34], and affects the total amount of food intake per day. CCK is secreted from I-type enteroendocrine cells in the duodenum and small intestine to intestinal lamina propria where it binds to CCK receptors on the vagus nerve terminal, transferring satiety signals to the hypothalamus via the brainstem and pontine parabrachial nucleus [34]. There are two different subtypes of CCK receptors, CCK-A and CCK-B. CCK-A is primarily expressed in the GI tract, while CCK-B is predominant in the CNS [35]. Otsuka Long-Evans Tokushima Fatty rats, an animal model of obese type 2 diabetes, have mutations in CCK-A [36].
Pancreatic polypeptide (PP)
Meal intake induces PP secretion from pancreatic islet PP cells via a vagal-mediated mechanism. A rise in circulating PP levels following a meal is in proportion to the calorific load and lasts for up to 6 hours [37]. Acute and chronic peripheral administration of PP reduces food intake in mice [38]. These anorectic effects of PP are thought to be mediated via the Y4 receptor in the brainstem and hypothalamus [38].
In humans, anorexigenic effects of PP persisted for 24 hours post-infusion, suggesting that PP may be involved in longer-term control of appetite [39]. Plasma PP levels were shown to be lower in obese subjects [40]. Interestingly, both basal and postprandial release of PP was reduced in patients with Prader-Willi syndrome, suggesting that defective PP secretion may account for hyperphagia in obese patients [41].
Peptide tyrosine-tyrosine (PYY)
PYY is secreted postprandially from the L cells of the ileum, colon, and rectum as a form of PYY1-36 [42], which is rapidly metabolized to PYY3-36 by the dipeptidyl peptidase (DPP)-4 in circulation. Circulating PYY3-36 binds to the Y2 receptor on presynaptic terminals of hypothalamic NPY/AGRP neurons with a high affinity [43], which results in inactivation of NPY/AgRP-producing neurons and induction of anorexia. Infusion of PYY3-36 in humans reduced consumption of food during test meals [44]. Obese subjects had lower plasma PYY3-36 levels compared to lean subjects [44]. Therefore, it has been suggested that reduced PYY secretion in the postprandial period may contribute to impaired satiety generation and development of obesity. Interestingly, the polymorphism of the PYY gene (Q62P), which impaired binding of PYY to the Y2 receptor, was associated with higher body weight [45].
GLP-1
GLP-1 is produced from a large precursor peptide preproglucagon in L cells of the ileum and colon. GLP-1 is secreted to systemic circulation where it is rapidly inactivated by DPP-4 [46]. Thus, the half-life of plasma GLP-1 is about 1 to 2 minutes. According to a recent meta-analysis [47], intravenous infusion of GLP-1 induced a reduction in food intake in both lean and obese humans with a lower effect in obese subjects. GLP-1 exerts anorexigenic effects through the GLP-1 receptor (GLP-1R), which is widely distributed in the brain, GI tract, and pancreas [48]. Administration of exendin-4, a DPP-4 resistant long-acting GLP-1R agonist, suppresses food intake in humans and rodents [49]. In addition to anorexigenic action, GLP-1 stimulates glucose-dependent insulin secretion by acting on pancreatic β-cells. Thus, DPP-4 inhibitors and degradation-resistant GLP-1 analogues are now used for the treatment of obese type 2 diabetes.
Oxyntomodulin (OXM)
OXM is produced from preproglucagon along with GLP-1 in intestinal L cells and has modest anorexigenic actions in rodents and humans [50]. The anorexic effects of OXM were antagonized by coadministration of GLP-1R antagonist and abolished in GLP-1R null mice [51], suggesting that OXM signals anorexia through GLP-1R.
Ghrelin
Ghrelin is a unique gut hormone in that it has an orexigenic effect. It was originally isolated from the rat stomach as an endogenous ligand of the growth hormone secretagogue receptor (GHS-R) and has been shown to have a GH-releasing effect [52]. Subsequently, ghrelin was identified as orexigenic hormone. Ghrelin administration stimulates food intake and body weight gain when administered centrally and peripherally [52]. Moreover, plasma ghrelin concentrations are elevated during a fast. Thus, ghrelin is considered to be a physiological hunger hormone. Of note, plasma ghrelin concentrations display a circadian rhythm: a rise before each meal and a rapid fall after eating, supporting a role for ghrelin in meal initiation. Fasting morning ghrelin concentrations have a negative correlation with fat mass index. Obese subjects displayed lower ghrelin levels compared with lean subjects [53]. Diet-induced weight loss in obese individuals increased plasma ghrelin levels [54]. These findings suggest that plasma ghrelin levels may represent a compensatory response to altered energy metabolism.
GI endocannabinoids system
The central and peripheral endogenous cannabinoid system appears to play a role in feeding regulation. Endocannabinoid receptors, CB1 and CB2, are expressed in the GI tract [55]. Administration of CB1 agonist increased food intake and reduced gastric motility [56]. Conversely, administration of a selective CB1 antagonist suppressed food intake and weight gain in obese animals [56], suggesting that endogenous endocannabinoids may have an orexigenic effect. Consistently, fasting elevated CB1 expression in the vagus and levels of endogenous CB1 ligand anandamide in the small intestine [56].
NUTRIENTS-RELATED SIGNALS
In addition to hormones, nutrients by themselves can relay satiety signals to the hypothalamus. Glucose signals satiety by acting on the hypothalamic glucose responsive neurons in the ARC and VMH [10] that have glucose-sensing machinery such as glucose transporter-2, glucokinase, and ATP-dependent potassium (KATP) channel as in pancreatic β-cells. Likewise, exogenous free fatty acids have an anorexigenic effect which is mediated through KATP channels [57]. In the hypothalamic neurons, fatty acid intermediates malonyl CoA and long chain fatty acyl-CoA are shown to signal satiety [58]. In the gut, oleoylethanolamide is released after a meal and generates satiety signals through the G-protein coupled receptor GPR119 [59]. In addition, the amino acid leucine can induce satiety by activating the mTOR and S6K signaling pathway in hypothalamic neurons [60].
CONCLUSIONS
We briefly illustrated the physiological mechanisms of appetite regulation with a focus on appetite regulators derived from the periphery (Fig. 2). In the CNS, the hypothalamus and brainstem play a central role in appetite regulation. Defective satiety generation in these areas leads to overeating and progression of obesity, although detailed mechanisms for this phenomenon are not completely understood. In addition, the recent epidemic of obesity is also associated with hedonic feeding. Therefore, future studies are needed to understand the modes of interaction between the metabolic center (hypothalamus, brain stem) and reward center (VTA, NA, forebrain) under normal-weight and obese conditions.
In recent decades, the gut has emerged as an important metabolic organ due to the fact that severe human obesity and combined metabolic disorders are successfully treated by bariatric surgery. Although changes in GLP-1, PYY, and ghrelin after bariatric surgery may explain a portion of the beneficial effects of nutritional bypass, the mechanisms of this phenomenon are largely unknown. Further research is needed to expand our understanding of the mechanisms of normal and abnormal regulation of food intake and eventually enable us to overcome obesity and its related metabolic disorders.
 XML Download
XML Download