

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
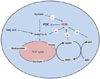
Selective fuel utilization depending on the fed-fast cycle is a crucial metabolic regulatory system in all mammals that is necessary to maintain a continuous and steady supply of ATP. In the fed state, increased availability of plasma glucose stimulates glucose oxidation and fatty acid synthesis. In the starved state, free fatty acids released from adipose tissue are selectively used for oxidative ATP generation in peripheral tissues and liver, and hepatic gluconeogenesis maintains the plasma glucose homeostasis. This regulation is mainly controlled by the activity of the pyruvate dehydrogenase complex (PDC), which regulates the entry of glycolytic products into the tricarboxylic acid cycle by catalyzing the oxidative decarboxylation of pyruvate to acetyl-CoA in mitochondria of mammalian cells. Although PDC is directly regulated by feedback inhibition by acetyl-CoA and NADH, both are end-products of oxidative decarboxylation of pyruvate and β-oxidation of free fatty acids, covalent modification of PDC via phosphorylation is recognized as critical for the long term regulation of PDC activity [1]. Pyruvate dehydrogenase kinase (PDK) and pyruvate dehydrogenase phosphatase (PDP) are key regulators of PDC activity that act in a phosphorylation-dephosphorylation manner. Thus, the opposing activities of PDK and PDP regulate PDC activity.
Four PDK isoenzymes (PDK1, 2, 3, and 4) are known to be expressed in a tissue-specific manner in mammals. Of these, PDK2 and PDK4 have attracted the most interest due to the finding that their expressions are increased in many tissues during starvation and in diabetes. PDK2 is the most widely expressed isoform and particularly enriched in the liver and kidney [2,3]. The expression of PDK4 is dominantly increased in oxidative skeletal muscle, heart, lactating mammary gland, and liver [3-5]. As PDC activity is primarily controlled by acetyl-CoA and NADH, the activities of PDKs are also affected by the products and substrates of the catalyzing reaction. The mitochondrial levels of acetyl-CoA, NADH, and ATP increases PDK activity, while the levels of pyruvate, CoA-SH, NAD+, and ADP inhibit its activity (Fig. 1). This system controls the acute regulation of PDK activity during short-term food intake-deprivation cycles. The activity of PDK in metabolic disorders such as diabetes, heart disease, fatty liver, and long-term starvation is mainly controlled by transcriptional up-regulation of PDK.
REGULATION OF PDK EXPRESSION BY INSULIN-FORKHEAD BOX O (FOXO) SIGNALING
PDK2 and PDK4 are highly expressed in liver, muscle, kidney, and heart tissue in starved and insulin-resistant animal models. Reduced insulin level or impaired insulin signaling contributes to the increase in both kinases. A remarkable increase in the amount of PDK4 mRNA occurs in the heart and skeletal muscle of streptozotocine-induced type 1 diabetic rat [4,6]. PDK4 is elevated in the skeletal muscle of insulin-resistant human subjects [7]. The negative regulatory effect of insulin on PDK4 expression was impaired in mice with acute insulin resistance induced by intra-lipid and lactate infusion, suggesting that insulin resistance may result in increase of PDK4 expression [8]. Likewise, insulin treatment reverse the increase of PDK4 and PDK2 expression in liver and skeletal muscle induced by dexamethasone [6,7].
The role of insulin in the regulation of PDK expression is mediated in large part by the transcriptional activity of FOXO proteins. In particular, FOXO1 is activated by starvation and prevents hypoglycemia by inducing the transcription of gluconeogenic genes in the liver, such as phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase), concurrent with PDK4 expression. To generate glucose, noncarbohydrate substrates such as pyruvate, lactate, and alanine must be conserved in the liver, which is dependent on the inactivation of PDC and a simultaneous increase in PDK activity. Fasting also activates FOXO1 in muscle, contributing to the induction of PDK and other genes involved in fatty acid utilization. Regulation of PDK by FOXO is reasonable to regulate fuel selection and maintain sufficient ATP levels through either glucose oxidation or fatty acid oxidation depending on the availability of energy source. Indeed, FOXO contributes to the regulation of PDK transcription by interacting with other nuclear hormone receptors such as the estrogen-related receptor (ERR), glucocorticoid receptor (GR), and peroxisome proliferator-activated receptor (PPAR). This is discussed in more detail below.
The human PDK4 gene possesses three insulin response sequences (IRSs) that are binding sites for FOXO1 and FOXO3 [9]. Overexpression of constitutively active FOXO1 and FOXO3, which contain alanines at the three protein kinase B (Akt/PKB) phosphorylation sites, augmented both basal and dexamethasone-mediated increased expression of PDK4 in HepG2 cells; these augmentations were eliminated by mutation of the IRSs in the PDK4 promoter [9]. In mouse muscle and C2C12 cells, nutrient deprivation induced FOXO gene expression, which mediated the up-regulation of PDK4 gene expression [10]. Nahle et al. [11] reported that over-expression of CD36 in C2C12 cells, which facilitates fatty acid uptake in muscle, induced FOXO1 expression, thereby contributing to the up-regulation of muscle PDK4 expression.
Although it is now well established that reduced insulin levels are responsible for the induction of PDK2, little is known about the PDK2 promoter. The insulin-mediated reduction of PDK2 mRNA expression in hepatoma cells might be associated with a decreased stability of the transcript [12]. We recently confirmed that PDK2 is dominantly expressed in the liver tissue of insulin-resistant animal models, including high fat dietinduced obese mice and db/db mice (unpublished data). Although the precise mechanism that mediates the up-regulation of PDK2 has not been elucidated, an increased fat supply in conjunction with insulin resistance may lead to an increased expression of PDK2 in the liver.
REGULATION OF PDKS BY NUCLEAR HORMONE RECEPTORS
PPARs
PPARs are a family of nuclear hormone receptors that function as transcription factors to regulate the expression of genes involved in metabolic pathways. Among the three main isoforms (PPARα, PPARβ/δ, and PPARγ), PPARα, an important adaptive regulator during prolonged fasting that promotes ketogenesis and fatty acid oxidation, is associated with increased expression of PDKs. Wu et al. [5] have reported a novel observation that WY-14,643, a PPAR-α agonist, dramatically induced both PDK4 mRNA and its protein in mouse skeletal muscle. Consequently, up-regulation of PDK4 by PPARα agonists, including fibrates and WY-14,643, has been investigated in several tissues such as heart, skeletal muscle, liver, and pancreatic β-cells [3,13-16]. Recent research shows that in skeletal muscle, pharmacologic activation of the liver X receptor (LXR) enhances PPARα-dependent up- and down-regulation of PDK4 in both fed and fasting conditions [17]. Although the clinical implication of LXR activation followed by a change in PDK4 expression, it has not been evaluated yet.
The results of a study performed in human and mouse kidney cells using the PPARβ/δ agonist GW501516 suggest that PPARβ/δ lies upstream of PDK2, PDK3, and PDK4 [18]. In addition, it has been reported that PPARβ/δ induces FOXO1 expression via transcriptional regulation in muscle cell lines [11]. PPARγ is highly expressed in adipose tissue and regulates adipogenesis and glucose metabolism. Treatment with the PPARγ agonist rosiglitazone increased the amount of PDK4 in white adipose tissue, but not in liver or muscle tissue [19]. This will be discussed in detail in the section REGULATION OF PDK4 IN ADIPOSE TISSUE. In HEK293 cells, PPARγ ligands influence the mRNA expression of PDK3 and PDK4, but not PDK2 [18]. In addition, some evidence exists to suggest that PDK2 is not likely a direct target of PPARs in metabolically active tissues [3,18,19].
It is of interest that free fatty acids and their derivatives have been implicated as endogenous PPAR ligands responsible for enhanced PDK expression in fasting and diabetes. Several reports demonstrate that long-chain fatty acids, such as palmitate and oleate, directly induce PDK4 mRNA in skeletal muscle and hepatoma cells [5,11,12]. Similarly, indirect supplies of fatty acids, such as increased levels of cardiac lipoprotein lipase and hepatic lipase, increase PDK4 mRNA expression by interacting with PPARα and PPARβ/δ [20,21]. When a hyperinsulinemic-euglycemic clamp was performed with healthy men, the suppression of PDK4 expression by insulin was attenuated by lipid infusion. In addition, this was independent of the activation of the Akt/PKB-mediated pathway, indicating that lipid accumulation is a more important factor than insulin signaling in the regulation of PDK4 expression in skeletal muscle [22]. On the contrary, insulin-mediated suppression of PDK4 expression in skeletal muscle was independent of its effect on plasma fatty acid levels, indicating that increased expression of PDK4 in starvation and diabetes may be due to insulin deficiency rather than to increased levels of fatty acids [23]. Further studies to identify the activators of PPARs responsible for regulation of PDK expression will provide insight into the role of PDK in metabolic disorders such as diabetes and insulin resistance.
GR
Dexamethasone treatment has been observed to markedly increase PDK4 expression in several tissues [12,24], suggesting that glucocorticoid is an important regulator of PDK4 expression in starvation and diabetes. A glucocorticoid response element (GRE) has been identified in the -824/-809 promoter region of the human PDK4 gene [9]. In addition, FOXO has been shown to participate in the induction of PDK4 by glucocorticoids via interaction with insulin response elements (IREs) located in a region near the GRE in the human PDK4 promoter [9]. Activation of protein kinase B-α, a component of intracellular insulin signaling, attenuated the dexamethasone-induced increase in PDK4 expression by interrupting the binding between FOXO and the IRE [9]. Insulin has also been shown to effectively decrease PDK4 expression augmented by dexamethasone in a similar manner [12]. Although dexamethasone treatment is reported to enlarge the lipoprotein lipase pool, as well as PDK4 expression, in the rat heart [24], whether the increased delivery of free fatty acids to the heart results in the augmentation of PDK4 expression is still controversial. Hyperglycemia, one of the clinical manifestations of Cushing syndrome, might be explained in part by the glucocorticoid-induced up-regulation of PDK4. It is not yet clear whether glucocorticoids regulate the expression of PDK2.
ERRs
ERRs are orphan nuclear receptors that are involved in the transcriptional regulation of cellular metabolic pathways. Of the three known isoforms, ERRα and ERRγ are expressed in metabolically active tissues such as skeletal muscle and liver. Several lines of evidence suggest that ERRα and ERRγ stimulate fatty acid oxidation through interaction with the PPARγ coactivator 1-α (PGC-1α) and gluconeogenesis via up-regulation of gluconeogenic genes such as PEPCK and G6P'ase, respectively. The roles played by ERRs in metabolic regulation suggest a relationship between ERRs and PDKs. ERRα has been shown to be involved in transcriptional activation of the PDK4 gene in C2C12 cells [25]. In that study, they showed that up-regulation of the PDK4 gene by ERRα was independent of PPARα, but was in partnership with PCG-1α. Up-regulation of PDK4 by ERRα was shown to be independent of PPARα, but dependent on a partnership with PCG-1α. Immediately following this study, the transcriptional regulation of PDK4 by ERRα and ERRγ in the liver was reported, which was associated with the recruitment of PCG-1α to the PDK4 promoter [26]. Both studies identified two ERR binding sites in the PDK4 promoter that are conserved in rat, mouse, and human PDK4 genes [25,26].
Thyroid hormone receptors (TRs)
The speculation that PDK is regulated by thyroid hormone (TH) arose from the observation that in hyperthyroidism, fatty acid oxidation is increased in various organs by altering the activity of CPT1, which is a major determinant of the rate of fatty acid oxidation [27,28]. In cultured hepatocytes and myocytes, triiodothyronine (T3) has been shown to enhance PDK activity [29,30]. Although the primary TH-responsive isoenzyme(s) in each tissue remain unknown, it is apparent that PDK2 and PDK4 are the predominant target genes of TH.
Previous studies have shown that experimental hyperthyroidism results in increased PDK activity with enhanced expression of PDK4, but not PDK2, in heart tissue [28,31]. In addition, another study has reported that the expression of PDK2 is increased in rat heart tissue in hypothyroid conditions, whereas acute T3 treatment paradoxically elevated PDK2 levels [32]. In the former experiments, they used relatively long-term hyperthyroidism models, but in the latter, T3 was infused for only 60 minutes. These study results suggest the possibility that different mechanisms mediate TH-dependent regulation of PDK in heart tissue depending on the duration of treatment.
In the liver, both PDK2 and PDK4 protein expression were enhanced in models of hyperthyroidism [3]. On the other hand, high fat diets, which increase the circulating lipid supply in a manner similar to hyperthyroidism, have been shown to enhance expression of PDK2 but not PDK4, in the liver, suggesting that up-regulation of PDK by TH in the liver might not be directly related to the supply of fatty acids. The transcriptional activity of TH is initiated by binding to the thyroid hormone receptor TR. The TR then binds to thyroid response elements (TRE) in conjunction with the recruitment of various coactivators. Two TREs in the rat PDK4 gene promoter have been identified, confirming that TH is able to regulate the transcription of PDK4 [33]. PGC-1α and CCAAT-enhancer-binding protein β (C/EBPβ) were discovered to be coactivators involved in the T3-mediated up-regulation of PDK4 [33,34]. Although the role of C/EBPβ has not been widely examined, it is well established that both TH and PGC-1α are linked to hepatic metabolism through the transcriptional regulation of gluconeogenic genes such as PEPCK. They also promote fatty acid oxidation, indicating that TH and PGC-1α have similar roles in the up-regulation of PDK [35,36]. Further study will be needed to determine the mechanism by which PDKs affect hepatic glucose-lipid metabolism in cooperation with transcriptional regulators, including TH, PGC-1α, and C/EBPβ.
OTHER FACTORS THAT REGULATE PDK EXPRESSION
Growth hormone (GH)
As previously noted, fasting-induced hormones, such as glucocorticoids, have the potential to regulate PDK expression. GH is also a counter-regulatory hormone that is induced during fasting and participates in preventing hypoglycemia. The first study to describe the relationship between GH and PDK was published in 1974, and it showed that a GH polypeptide inhibited PDC activity in muscle [37]. However, subsequent data demonstrating that GH could increase PDC activity in rat hepatocytes, human mononuclear cells, and rat myocardium [38-40] were in contrast with recent studies that demonstrated that GH up-regulated PDK4 expression [41,42]. In one study, insulin could attenuate the ability of GH to induce PDK4 mRNA expression in adipose tissue, which is to be expected considering that GH plays roles as a counter-regulatory hormone in glucose metabolism. In addition, up-regulation of PDK4 by GH was mediated by STAT5A, which is a well-known downstream target of GH. It was recently established that GH also induces mRNA expression of PDK4 in the liver in a STAT5-dependent manner [42]. This induction was inhibited by treatment with metformin, which was accompanied by AMPK-mediated SHP activation. This novel observation of a GHSTAT5-PDK4 cascade in the liver may have clinical implications for the treatment of conditions with dysregulated hepatic metabolism associated with excess levels of GH, such as acromegaly.
Krüppel-like factor 15 (KLF15)
KLF15 is increased by glucocorticoid signaling during fasting and decreased by insulin signaling during feeding in hepatocytes. The enhanced expression during starvation is associated with its ability to increase the expression of gluconeogenic genes such as PEKCK. In this regard, we hypothesized that KLF15 would interfere with the transcription of PDK4 in liver tissue. As we had speculated, PDK4 promoter had the binding consensus motif for KFL15, and KLF15 could induce the mRNA expression of PDK4 in the liver cell line (unpublished data). This observation was confirmed by demonstrating that a mutation in the promoter region at the presumptive KFL15 binding site affected the expression of PDK4.
Adiponectin and AMPK
Adiponectin, which has been recognized as an insulin sensitizer in skeletal muscle, stimulates fat oxidation via the activation of AMPK. McAinch and Cameron-Smith [43] investigated the effect of adiponectin and AMPK activation on PDK4 gene expression in cultured skeletal muscle tissue from lean, obese, and obese diabetic humans. Both adiponectin and pharmacological activation of AMPK by 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR) decreased PDK4 expression in obese and diabetic samples, suggesting the possibility that the decrease in blood glucose mediated by adiponectin resulted from the down-regulation of PDK4 in skeletal muscle. Meanwhile, it was published that AMPK activation by hypoxia or AICAR with free fatty acid supply synergistically induced PDK4 [13].
REGULATION OF PDK4 IN ADIPOSE TISSUE
Compared to other metabolic regulatory organs, such as skeletal muscle, liver, and heart, a relatively small number of studies on the expression and regulatory mechanism of PDK in adipose tissue have been published. Triglyceride turnover in adipose tissue is a crucial metabolic process because it facilitates fatty acid utilization during fasting. Furthermore, the release of excessive amounts of plasma free fatty acids from adipose tissue is associated with insulin resistance [44]. These effects can be mediated via glyceroneogenesis, which occurs through the conversion of non-glucose molecules such as pyruvate to glycerol. This pathway begins with the conversion of pyruvate to oxaloacetate by pyruvate carboxylase (PC) in the mitochondria, suggesting that the ratio of PDC to PC activity is a key regulator of this pathway. It has been suggested that changes in PDC activity mediated by PDK4 expression could affect glyceroneogenesis in adipose tissue. Cadoudal and colleagues [19] showed that rosiglitazone increased PDK4 expression in a transcriptionally regulated manner in adipose tissue in an insulin-resistant animal model and in cultured adipocytes from humans and rats. In addition, they provided evidence that induced PDK4 played a role in white adipose tissue glyceroneogenesis, demonstrating that pyruvate is highly incorporated into neutral lipids by rosiglitazone and is reduced by both PDK inhibitors and siRNA of PDK4.
Epinephrine has also been used to activate PDK4 in adipose tissue. Wan et al. [45] demonstrated that acute epinephrine treatment increased PDK4 mRNA in cultured adipose tissue and epididymal fat in rats, and that this increase was attenuated by blocking p38 mitogen-activated protein kinase activity. Similarly, a 2-hour swim exercise also leads to an increase in PDK4 mRNA levels in epididymal adipose tissue. They performed this experiment with obese, insulin-resistant rats [46]. Interestingly, the induction of PDK4 in adipose tissue by acute epinephrine injection was to a similar extent in lean and high fat diet (HFD) rats, whereas the activation of p38 was blunted in adipose tissue only in HFD rats. These findings demonstrated that increased PDK4 expression in adipose tissue induced by epinephrine or exercise is independent of p38. Although the transcription factor involved in PDK4 up-regulation by rosiglitazone or epinephrine remains unknown, these results indicate that PDK4 may play a pivotal role in regulating glucose-lipid metabolism in adipose tissue.
CONCLUSIONS
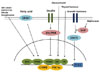
Exploring the transcriptional pathways involved in regulating PDK isoenzymes expression will lead to a better understanding of the molecular mechanisms that underlie the development of metabolic disease such as diabetes. This review summarizes recent progress in elucidating the transcriptional control of PDK, focusing on the PDK2 and PDK4 isoenzymes that play a role in metabolic pathways in insulin-responsive tissues (Fig. 2). Insulin and its downstream targets, such as FOXO, are major regulators of PDK expression. In addition, several nuclear receptors and hormones that also function in metabolic pathways coordinately participate in the transcriptional regulation of PDK.
Studies that evaluate the efficacy of controlling PDC activity by down-regulating PDK expression with PDK inhibitors in animal models of metabolic disease, including diabetes, fatty liver, and obesity, will provide an opportunity to identify new therapeutic targets for the treatment of metabolic diseases.
 XML Download
XML Download