

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Diabetes diagnoses are increasing at an alarming rate worldwide. The majority of diabetes-related deaths arise from vascular complications such as myocardial infarction, cerebrovascular disease, and peripheral vascular disease [1]. Large prospective randomized clinical studies show that long-term glycemic control is an important predictor of diabetic vascular complications [1,2].
There is considerable evidence that many of the biochemical pathways adversely affected by hyperglycemia including glucose oxidation, the formation of advanced glycation end-products (AGE), and activation of polyol pathways, are associated with the generation of reactive oxygen species (ROS), ultimately leading to increased oxidative stress in a variety of tissues [3]. In the absence of an appropriate compensation by the endogenous antioxidant defense network, increased oxidative stress leads to the activation of stress-sensitive intracellular signaling pathways and the formation of gene products that cause cellular damage and contribute to the late complications of diabetes [3,4].
Major reactive species, including ROS and reactive nitrogen species (RNS) consist of paramagnetic free radicals (superoxide O2•-, hydroxyl HO•, peroxy ROO•, and nitric oxide free radical NO•) and diamagnetic molecules (hydrogen peroxide H2O2 and peroxynitrite ONOO-) which are products of the reactions of these free radicals. All these species were previously considered to be toxic agents capable of damaging biomolecules. However it is now known that physiological free radicals superoxide and nitric oxide are relatively harmless species that are able to initiate or mediate many enzyme- and gene-dependent reactions in both physiological and pathophysiological processes. Increasing evidence in experimental and clinical studies suggests that ROS/RNS play important roles in the pathogenesis of diabetic vascular diseases [4].
This review summarizes recent knowledge on the relevance of oxidative and nitrosative stress in the pathophysiology of diabetic vascular complications.
REACTIVE OXYGEN SPECIES SIGNALING IN DIABETES
Sources of ROS
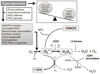
As the development of diabetes mellitus is characterized by high serum glucose levels, this prooxidant molecule can be an origin of ROS overproduction (Fig. 1). High glucose (HG) levels can initiate the production of superoxide and hydrogen peroxide, precursors of reactive free radicals, which are able to stimulate the decline of antioxidant systems, directly damage many biomolecules, and increase lipid peroxidation in diabetes [5,6].
The most important sources of ROS under hyperglycemic conditions are mitochondria and NAD(P)H oxidases. Hyperglycemia decreases glyceraldehyde-3-phosphate dehydrogenase (GAPDH) activity in bovine aortic endothelial cells via the increased production of mitochondrial superoxide and a concomitant increase in hexosamine pathway activity [7,8]. Mitochondrial ROS have also been implicated in diabetic complications and progression of the underlying diabetic state; however, it is not clear whether mitochondria of diabetic origin actually generate ROS independent of the surrounding diabetic milieu.
NAD(P)H oxidases are another important ROS source in diabetes. Superoxide production significantly increases by upregulated NAD(P)H oxidase and endothelial NO synthase in human blood vessels from subjects with type 2 diabetes [9]. These effects of NAD(P)H oxidase and uncoupling eNOS might be mediated by protein kinase C (PKC) signaling. Our group assumed that the enhanced proliferative capacity of diabetic vascular smooth muscle cells (VSMC) is a consequence of NAD(P)H oxidase activation stimulated by the activation of a protein tyrosine kinase [10]. It has been suggested that ROS formation by HG-stimulated NAD(P)H oxidase can also upregulate antioxidant enzymes [11]. Therefore NAD(P)H oxidase might be a double-edged sword, providing feedback defense against excessive ROS generation through the activation of receptor tyrosine kinases and the redox-sensitive transcription factor Nrf2-Keap1 signaling pathway [11]. It is of importance that Block et al. [12] have recently identified NAD(P)H oxidase (Nox4) as mitochondrial cytochrome c oxidase subunit IV as being an additional source of ROS in diabetes. It should be noted that xanthine oxidase is also a potential source of ROS formation in diabetes.
ROS overproduction in diabetes
Diabetes causes increased oxidative stress in various tissues as evidenced by increased levels of oxidized DNA, proteins, and lipids. Besides damaging the functions of these molecules, oxidative stress also triggers a series of cellular responses, including the activation of PKC, NF-κB, and JNK stress-associated kinases [13]. Inappropriate activation of these important regulatory molecules can have deleterious effects on cellular functions, and is thought to contribute to the pathogenesis of various diabetic vascular complications. However, it is not clear how hyperglycemia leads to increased oxidative stress. Most likely, both the increased production of ROS and decreased capacity of the cellular antioxidant defense system combine to produce oxidative stress in diabetes.
Hyperglycemia increases oxidant production by multiple pathways rather than a single dominant pathway. Glucose can undergo non-enzymatic reactions to form gluco-oxidants and glycated products, which can be oxidants [13]. Metabolism of excessive intracellular glucose can occur by several processes such as aldose reductase, mitochondrial oxidative phosphorylation, activation of NAD(P)H oxidases, and alteration of NADPH/NADP ratios [3]. Among these possibilities, recent work has centered on mitochondrial metabolism and activation of NAD(P)H oxidases [3,13]. It has been suggested that most glucose-induced oxidants are derived from overproduction of superoxide by the mitochondrial electron-transport chain [3]. By-products of this process will trigger signaling cascades such as activation of PKC, production of hexosamine, increased flux via aldose reductase, and production of glycated products. However, other investigators have reported that high glucose levels can activate NAD(P)H oxidases in vascular cells independent of mitochondrial metabolism [14]. It is known that the ROS activation of PKC stimulates subsequent ROS overproduction; therefore this enzymatic cascade can be an important harmful signaling process in hyperglycemia and diabetes. Thus, Koya and King [15] suggested that glucose's adverse effects in diabetes and hyperglycemia might be a consequence of ROS-stimulated activation of the diacylglycerol (DAG)/PKC enzymatic pathway because treatment with α-tocopherol prevented glucose-induced vascular dysfunctions and inhibited DAG/PKC activation. ROS signaling is also an important stimulus of harmful enzymatic cascades catalyzed by protein kinase Akt/B [16].
Recently, two other aspects have been found to be related to glucose-induced oxidative stress. First, several studies have suggested that intermittent low and high glucose conditions (a condition known as "glucose variability") are even more deleterious to endothelial cell function than a steady, constant increase in glucose [17]. These conditions also induce endothelial cells to enter into a proinflammatory state, and this state is associated with the upregulation of various adhesion molecules and proinflammatory cytokines [17]. Some of the pathways implicated in these exacerbated cellular responses, involve activation of PKC, NAD(P)H oxidases, and mitochondrial oxidants. Secondly, it has been shown in culture and in diabetic rats that endothelial cells exhibit a persistence or 'memory' of induced basement membrane mRNA expression long after normalization of high glucose levels, a condition called "glucose memory" [18]. This finding suggests that glucose induces long-lasting deleterious effects that persist beyond the period of hyperglycemia.
Abundant evidence exists that many protein, lipid, and DNA markers of oxidative stress are increased in cultured vascular cells exposed to high glucose levels and in vascular tissues from animals and human subjects with diabetes [13]. In cultured vascular cells, elevating glucose levels in the media increases the production of oxidants such as gluco-oxidants, glycated compounds, oxidized low density lipoprotein (LDL), superoxide, and nitrotyrosine [14]. Similarly, elevated levels of isoprostanes, 8-hydroxydeoxyguanosine, and lipid peroxides have been reported in diabetic animals and in human subjects with diabetes [14]. Although numerous reports have substantiated that oxidant production is increased in diabetes, clinical evidence for tissue damage as a result of oxidative stress has not been clearly demonstrated because both plasma and cells contain a large reserve of antioxidants. In fact, the levels of various antioxidants in plasma and cells have not consistently been shown to decrease in diabetic states [14].
Role of ROS in development of macrovascular complications
Abnormalities in endothelial and vascular smooth muscle cell function, as well as the coagulation system, contribute to vascular complications in diabetes.
Vascular endothelial dysfunction
Endothelial cells modulate vascular function and structure. In normal endothelial cells, nitric oxide is synthesized and released to maintain vascular homeostasis. Clinical studies have consistently found that endothelium-dependent vasodilation is abnormal in subjects with diabetes [15]. The major contributor to endothelial oxidative stress is the increased production of superoxide. This seems to occur via two principal sources: NAD(P)H oxidases and uncoupled endothelial nitric oxide synthase (eNOS). Hyperglycemia, AGE, free fatty acid (FFA), and oxidized LDL have been shown to increase endothelial NAD(P)H oxidase activity and the activation of NAD(P)H oxidases by hyperglycemia and FFA has been shown to be mediated by PKC [16]. Vessels isolated from diabetic subjects exhibit increased superoxide production that is inhibited by diphenylene iodinium, and demonstrate increased expression of several NAD(P)H oxidase subunits (p22phox, p47phox, and p67phox) [17], suggesting that NAD(P)H oxidases are more active in diabetes. Not only does excess superoxide itself cause increased oxidative stress, but it can also react with nitric oxide (NO•) to produce peroxynitrite [18], which, in turn, can oxidize tetrahydrobiopterin (BH4), thus reducing its availability to eNOS [17,18]. In the presence of reduced concentrations of BH4, eNOS becomes uncoupled and transfers electrons to molecular oxygen instead of L-arginine to produce superoxide rather than NO•. The presence of uncoupled eNOS in the diabetic vasculature is supported by a study in which diabetic vessels were found to produce less superoxide when incubated with the NO• synthase inhibitor NG-nitro-L-arginine methyl ester [17]. BH4 availability may also be decreased by a reduction of its synthesis. The expression of GTP-cyclohydrolase I (GTPCH), the rate-limiting enzyme for de novo BH4 synthesis, is reduced in diabetic rats [19]. Furthermore, transgenic mice overexpressing GTPCH treated with streptozotocin (STZ) are able to maintain endothelial function [19]. Clinical studies have demonstrated that BH4 supplementation given to diabetic subjects improves their endothelium-dependent vasodilation, indicating that uncoupled eNOS plays a role in diabetic endothelial dysfunction [17-19].
An additional effect of high serum glucose levels is delayed replication of large-vessel endothelium. Superoxide dismutase, catalase, and reduced glutathione protect human endothelial cells from glucose-induced delay in replication, offering further evidence of the importance of oxidative stress in diabetes [20].
Vascular smooth muscle dysfunction
In type 2 diabetic subjects, the vasodilator response to endogenous nitric oxide donors is diminished [15,20], suggesting that there is a fundamental abnormality in VSMC function. The oxidative stress produced in VSMC in diabetes may shift them from a contractile to a proliferative phenotype, thus further inhibiting vasodilation and enhancing lesion formation. Subjects with diabetes have increased proliferation and migration of VSMC into atherosclerotic lesions [21]. There are a number of studies showing that exposure of VSMC to high glucose conditions results in oxidative stress and subsequent cell proliferation.
Hyperglycemia causes PKC activation and subsequent ROS production via NAD(P)H oxidase in cultured aortic smooth muscle cells [14]. In STZ-treated rats, a p22phox-containing NAD(P)H oxidase was found to be a mediator of VSMC proliferation [22]. Additionally, the polyol pathway has been implicated in hyperglycemia-induced, PKC-directed NF-κB activation, as inhibition of aldose reductase is able to mitigate both PKC and NF-κB activation in cultured rat aortic smooth muscle cells [23]. Despite the increased proliferation and migration of VSMC in subjects with diabetes [21], atherosclerotic lesions from these patients have fewer VSMC, suggesting that VSMC death potentially plays a role in plaque instability and subsequent rupture [24]. A study in human aortic smooth muscle cells revealed that high glucose conditions cause cell necrosis via hydrogen peroxide [24]. Additionally, oxidized LDL is able to induce VSMC apoptosis via ROS and may be increased in diabetic subjects [24].
Role of ROS in development of microvascular complications
The microvascular complications of diabetes, including nephropathy, retinopathy and neuropathy, are common manifestations of diabetes. Although the mechanisms underlying the development of these conditions are incompletely understood, oxidative stress has been implicated. In isolated kidneys, exposure to ROS causes a drastic dose-dependent decrease in de novo synthesis of heparan sulfate, an effect that is reversed by catalase [25]. In addition, the ability of glomeruli isolated from STZ-induced diabetic rats to degrade hydrogen peroxide is greatly impaired; this has been attributed to either decreased catalase activity or altered glutathione redox cycling [25]. Recent data has shown that the expression of the NAD(P)H oxidase subunits Nox4 and p22phox are upregulated in the kidney of STZ-induced diabetic rats and that NAD(P)H oxidase-dependent production of ROS may cause DNA damage in diabetic renal tissues leading to the development of nephropathy [26].
Evidence for the participation of oxidative stress in the pathogenesis of diabetic retinopathy is scant and limited to studies published in abstract form. One report showed that the activity of NAD(P)H oxidase was increased in the retina of diabetic rats and that this might be involved in the development of diabetic retinopathy [27].
Stronger evidence exists for an involvement of ROS in the etiology of early experimental diabetic neuropathy. One report showed that probucol treatment prevents the reduction in nerve conduction velocity in STZ-induced diabetic rats and in normal rats treated with primaquine. The authors suggested that nerve dysfunction depends on oxidative stress that causes neurovascular defects resulting in endoneural hypoxia [28]. Treatment with the transition-metal chelating agents deferoxamine and trientine, which can prevent auto-oxidation, corrected nerve conduction and blood flow changes that occurred 2 months after the induction of diabetes in this model [28].
RNS SIGNALING IN DIABETES
Nitric oxide is an endothelium-derived relaxing factor (EDRF) and its role is very important in the normal activity of cells. Therefore, it might be expected that as in other pathological states a decrease in nitric oxide can take place in diabetes. On the other hand, nitric oxide is soluble in aqueous and lipid media and it rapidly diffuses through the cytoplasm and plasma membranes. In an inflammatory state, such as obesity, the immune system produces both superoxide and nitric oxide, which may react together to produce significant amounts of peroxynitrite anion (ONOO-). Peroxynitrite is a potent oxidizing agent that can cause DNA fragmentation and lipid peroxidation [29]. There are multiple lines of evidence demonstrating the formation of peroxynitrite in diabetic vasculature, both in experimental models and in humans [30,31]. The tissues and species in which peroxynitrite has been identified in experimental animals and in humans include plasma, kidney, blood vessels, retina, heart and peripheral nerves and have been overviewed recently [30]. The mechanisms that underlie peroxynitrite-induced diabetic complications and vascular alterations are multiple [31,32]. One of the important pathways of peroxynitrite-mediated vascular dysfunction in diabetes involves activation of the nuclear enzyme poly(ADP-ribose) polymerases (PARP enzymes). Activated PARP-1 cleaves NAD+ into nicotinamide and ADPribose and polymerizes the latter on nuclear acceptor proteins. Peroxynitrite-induced overactivation of PARP consumes NAD+ and consequently ATP culminating in cell dysfunction, apoptosis or necrosis [33]. Thus Langenstroer and Pieper [34] found that spontaneous EDRF released in diabetic rat aorta can be unmasked by the addition of superoxide dismutase (SOD) because SOD produces a significantly greater relaxation in diabetic aorta compared to control aorta. It means that NO synthases in diabetes produce superoxide in an uncoupled state. Other authors have also suggested that peroxynitrite formed by uncoupled NO synthases in human blood vessels from subjects with diabetes type 2 can be a mediator of the cytotoxic effects of high glucose [35].
TRIALS OF ANTIOXIDANT THERAPY FOR DIABETIC VASCULAR COMPLICATIONS
Many trials in diabetic subjects and animal models of diabetes have attempted to determine whether antioxidant treatments can prevent or delay the onset of diabetic complications.
If the effects of ROS/RNS signaling in enzyme/gene processes depend on the levels of ROS/RNS production, then ROS/RNS-induced damage in diabetes as well in the other pathologies such as cardiovascular diseases, inflammation, and aging cannot be successfully inhibited by the traditional antioxidants vitamin C or vitamin E which are unable to react with superoxide [36,37]. Nonetheless some aforementioned data demonstrate the successful application of various antioxidants and free radical scavengers for suppression of damaging processes in diabetes.
Experimental animal trials
Several antioxidants have been studied in diabetic and insulin-resistant animals, mostly rodents, with respect to their ability to correct vascular and neurologic dysfunction [28].
α-Lipoic acid is needed to regenerate glutathione and oxidized vitamins C and E. In animal studies, vitamins C and E and α-lipoic acid improve nerve conduction velocity and blood flow to the peripheral nerves, reduce retinal leukocyte adhesions, and improve cataract formation, suggesting that they may be an effective treatment of diabetic complications [28,38]. Furthermore, vitamins C and E, either individually or in combination, normalize many parameters of oxidative stress such as lipid peroxidation, isoprostane production, plasma malondialdehyde, and NF-κB activation in diabetic animals [39]. Besides biochemical changes, many early or functional markers of diabetic retinopathy, nephropathy, neuropathy, and even cardiovascular disease have been reported to be prevented or reversed by antioxidants, including blood flow, nerve conduction velocity, permeability, endothelial dysfunction, albuminuria, and vascular contractility [38]. A few reports have indicated that vitamins C and E may even prevent late pathological changes in the retina and peripheral nerves of diabetic animals [39].
PKC inhibitors may also function as antioxidants via their inhibitory effect on hyperglycemia-induced activation of NAD(P)H oxidase. These experiments only indirectly implicate oxidative stress, however, since PKC likely regulates diabetic complications on multiple levels. Oral administration of roboxistaurin (LY333531), a specific PKC-β inhibitor, to diabetic rats prevents increased albumin excretion, elevated glomerular filtration, and abnormal retinal hemodynamics [14]. Consistent with this, one report showed that angiotensin converting enzyme (ACE) inhibitors and angiotensin receptor blockers prevent the development of albuminuria in STZ-induced diabetic rats in parallel with prevention of increased expression of the p47phox component of NAD(P)H oxidase and production of its oxidative products [40].
Human clinical trials
Most clinical studies on the effects of antioxidant treatments have used early surrogate markers and are limited in duration and sample size.
A small, short-term single blind study showed that administration of an antioxidant mixture (N-acetylcysteine, vitamin E, and vitamin C) to control subjects, as well as subjects with type 2 diabetes and impaired glucose tolerance reduced the increase in oxidants and endothelial markers evoked by a fatty meal [41]. Another, short-term, but randomized double blind, placebo controlled study examined the effects of vitamins E and C with or without zinc and magnesium and demonstrated reduced albumin excretion, along with evidence of reduced lipid peroxidation in groups receiving the vitamins [42]. Although many clinical trials with such antioxidant vitamins investigating the progression of diabetic complications conclude as negative or inconclusive, one may wish to see additional studies, perhaps involving a combination of these vitamins, or mega-dose therapies.
Studies using α-lipoic acid suggest that it may improve the symptoms of diabetic polyneuropathy [13]. α-lipoic acid has the additional property of increasing glucose transport in muscle cells, which may be related to its antioxidant properties [13]. Clinical trials are now ongoing to assess the effects of PKC-β inhibition on diabetic retinopathy and neuropathy. The beneficial effects of a PKC-β specific inhibitor might also partly due to its inhibition of ROS production [14]. Further investigation into the antioxidant properties of this agent is necessary.
Importantly, pharmacologic agents currently in use that have been shown to be effective in reducing cardiovascular mortality are known to have antioxidant properties. ACE inhibitors and HMG-CoA reductase inhibitors or statins have already demonstrated beneficial effects on diabetic subjects in large randomized controlled-trials [43]. Interestingly, ACE inhibitors, which act partially to prevent the pro-oxidant effects of angiotensin II, were shown to prevent the onset of type 2 diabetes in the Heart Outcomes Prevention Evaluation Study [43]. Statins have been shown to exert vascular antioxidative properties [44].
Recent approaches to antioxidant therapy
The use of new antioxidant agents that penetrate specific cellular compartments may provide a new approach to dealing with oxidative stress in diabetes. Agents such as idebenone andmito-Q, which are selectively taken up into mitochondria should be searched further [45,46]. Edaravone, 3-methyl-1-phenyl-2-pyrazolin-5-one, is a novel scavenger of free radicals [47] and has been shown to ameliorate renal ischemia/reperfusion injury by scavenging free radicals produced in renal tubular cells and inhibiting lipid peroxidation [48]. Mimetics of superoxide dismutase have been investigated in experimental models of diabetes. Tempol (4-hydroxy-2, 2, 6, 6-tetramethylpiperidine 1-oxyl) improved endothelial function in Zucker diabetic fatty rats and in db/db mice [49]. As NAD(P)H oxidase is an important source of reactive oxygen species, targeting this enzyme may provide another approach to reducing the oxidative stress of diabetes and its consequences. Indeed, administration of the NAD(P)H oxidase assembly inhibitor apocynin attenuated the long-term effects of streptozotocin-induced diabetes in producing albuminuria and glomerulosclerosis [50]. Resveratrol is a polyphenol phytoalexin that can reduce vascular superoxide levels [51] and restore endothelial function in type 2 diabetes by inhibiting tumor necrosis factor-alpha induced activation of NAD(P)H oxidase and preserving eNOS phosphorylation [52]. NF-E2-related factor-2 (Nrf2) is a transactivator of genes that contain an antioxidant response element in their promoter. Such genes code for a number of enzymes including NADPH:quinone oxidoreductase, glutathione S-transferases and aldo-keto reductases, that have an important role in the protection of cells against oxidative stress [52].
CONCLUSIONS
The present paper is a review of published studies on the role of ROS/RNS in the pathogenesis of diabetic vascular complications. A large number of studies indicate that oxidant production is clearly increased through multiple pathways as a result of hyperglycemia. It has recently been suggested that diabetic subjects with vascular complications may have a defective cellular antioxidant response against the oxidative and nitrosative stress generated by hyperglycemia. This raises the concept that antioxidant therapy may be of great benefit to these subjects. However, supportive evidence that antioxidants can provide beneficial effects on diabetic vascular complications in large clinical studies is lacking, largely due to the lack of a suitable antioxidant supplement. Further initiatives to study the mechanisms that cause the generation of oxidative and nitrosative stress in diabetes may lead to the discovery and evaluation of new antioxidant molecules that could inhibit the diabetes-related oxidative and nitrosative stress mechanisms.
 XML Download
XML Download