

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Diabetes mellitus is one of the many chronic diseases that has a markedly increasing rate of prevalence. In Korea, the prevalence of diabetes reached almost 10% in 2009 in subjects older than 20 years of age [1]. Type 1 diabetes is an autoimmune disorder characterized by the destruction of pancreatic islets by immunologic insult and type 2 diabetes is caused by insulin resistance and decompensated insulin secretion from pancreatic islets. As these two main types of diabetes are increasing worldwide, vitamins have gained particular attention as environmental "agents of influence."
Vitamin A is an essential vitamin that must be derived from vitamin-A-rich foods as well as foods containing the carotenoid β-carotene, which is composed of 2 retinol molecules [2]. Retinoic acid (RA) is the active metabolite of vitamin A, and is a critical signaling molecule during the development of vertebrates [3]. Retinoids have long been appreciated as a crucial factor for controlling the differentiation program of certain epithelial cells and for their effects on vision, growth, reproduction and resistance to infection. Vitamin A deficiency could result in impaired cellular differentiation, reduced resistance to infection, anemia and ultimately, death. In a very old study by Wolbach and Howe [4] in 1925, when the rats were deprived of vitamin A from diet, epithelial growth was greatly augmented and they could see neoplasm-like growth in epithelial tissues. Conversely, many aquatic species (such as shark) exhibit a paucity of neoplasms because of their high content of vitamin A [5]. Currently, retinoic acid is officially accepted as the primary treatment for acute promyelocytic leukemia [6].
Although many studies are performed on the relationships between retinoic acid metabolism and embryogenesis and cancer, not many studies have looked at the relationship between retinoic acid and diabetes. Therefore, we want to briefly review the recent research in retinoic acid metabolism and its relationships with diabetes from the clinical and scientific perspectives.
OVERVIEW OF RETINOIC ACID METABOLISM
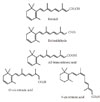
Analogs of retinol, with or without biological activity, are called retinoids. The only source of retinoids for most animals is diet as these compounds cannot be synthesized de novo. Diet (for example through milk, liver, and eggs) provides retinol as retinyl ester or a provitamin carotenoid (for example, in carrots and red pepper), of which β-carotene represents the most efficient precursor [2]. The predominant natural retinoid in circulation is retinol in micromolar levels, which is derived from the carotenoid, β-carotene or provitamin A, and retinyl ester, particularly palmitate, is the most abundant storage form, with liver serving as the main, but not the sole site of storage. The primary function of retinol and retinyl is to serve as the precursor for the biosynthesis of active retinoids, that is, the first initiator. Retinol has 6 biologically active isoforms: all-trans, 11-cis, 13-cis, 9,13-di-cis, 9-cis, and 11,13-di-cis, with all-trans being the predominant form (Fig. 1).
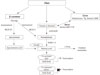
All-trans RA (atRA) is a small lipophilic molecule (300 daltons) that circulates in plasma and is bound to albumin at a concentration of 1 to 10 nmol/L [7,8]. Two retinoic acid (RA) isomers have been identified in vivo: 9-cis RA and 13-cis RA. The physiological function of 13-cis RA is unclear and it is detected at significantly lower concentrations in both mice and humans [9,10]. 9-cis RA binds with high affinity to a distinct type of retinoic receptor, the retinoid X receptor (RXR) [11,12]. AtRA is generated from the parental vitamin A molecule, all-trans-retinol, by two consecutive oxidation steps: oxidation from retinoid to retinal and then to RA (Fig. 2). These oxidation steps are mediated by retinol dehydrogenases and retinaldehyde dehydrogenases. Two classes of enzymes function as retinol dehydrogenases in vitro: medium-chain alcohol dehydrogenases and short-chain dehydrogenases/reductases (SDRs) [13]. AtRA and 9-cis-RA can regulate the expression of multiple genes [12], and as the transcriptional activities of RA can lead to differentiation, cell cycle arrest, and apoptosis, the compound often inhibits cell growth. Such activities enable RA to suppress carcinoma cell proliferation and provide the rationale for use in cancer treatment. RA is clinically used for treatment of malignancies (e.g., promyelocytic leukemia, Kaposi's sarcoma, and premalignancies) [14].
The transcriptional activities of RAs are mediated by several nuclear receptors (Fig. 2). Retinoic acid receptor (RAR) belongs to a large family of steroid hormone nuclear receptors and RARs α, β, and γ bind both atRA and 9-cis RA, whereas the RXRα, β, and γ bind to 9-cis RA. 13-cis RA is not a ligand for retinoid receptors, but can readily convert to retinoid receptor ligand. The mRNA expression of RARα, RXRα, and RXRβ is ubiquitous, whereas RARβ, γ, and RXRγ exhibit tissue-restricted patterns of expression [15,16]. RXRs regulate transcription as homodimers, and they also serve as obligatory heterodimerization partners for multiple other nuclear receptors within subclass I of the family, including RARs, peroxisome proliferator-activated receptors (PPARs), and the vitamin D receptor, thyroid hormone receptors and farnesoid X receptor, liver X receptor, and other key transcriptional sensors of nutrients and metabolites that help maintain homeostasis in metabolism and immune responses [17,18].
The transcriptional activation by RAR is dependent on the formation of the RAR/RXR heterodimer, which occurs on two interfaces, the ligand-binding domain (LBD) and DNA-binding domain (DBD). Mice null for RARα show some of the features of vitamin A deficiency with decreased viability, growth deficiency, and male sterility [19]. However, most of the defects of these mice could be reversed by RA treatment. Null mice for RARβ exhibit a selective loss of striosomal compartmentalization in the rostral striatum and also display locomotor defects, which are correlated with dopamine signaling, implicating retinoids in the regulation of brain function [20,21]. RARγ is highly expressed in the skin and RARγ-null mice display some defects associated with vitamin A deficiency, which can be rescued by RA treatment, indicating that RARγ mediates some of the retinoid functions in vivo [22,23].
Retinol is delivered to target tissue by the ligand-bound cellular retinol binding protein (CRBP) (Fig. 2), where retinol is degraded to produce retinoids through multiple enzymatic reactions [3]. In 1970, the cell surface receptor for retinol binding protein (RBP) was identified and since then, there has been accumulating evidence of the existence of the RBP receptor in various tissues [24]. This cell surface receptor binds to RBP and mediates retinol uptake from the ligand-bound CRBP. This cell surface receptor for RBP was shown to be stimulated by retinoic acid gene 6 (STRA6). STRA6 is a widely expressed multitransmembrane protein that is broadly expressed in murine embryo, but has more restricted expression in adults.
In target tissue, retinol either associates with CRBP or serves as a substrate for several cytosolic and microsomal enzymes termed retinol dehydrogenases (RDH) that oxidize retinol to retinaldehyde (Rald) [25,26]. Retinol dehydrogenases are members of the alcohol dehydrogenase (ADH) or SDR families. Retinol metabolism is catalyzed by ubiquitously expressed ADH3 as well as by the tissue-restricted ADH1 and ADH4. All three isoforms could oxidize all-trans retinol to all-trans retinaldehyde.
RA is produced from Rald through irreversible oxidation by retinaldehyde dehydrogenase (Raldh) (Fig. 2) [27]. It has been demonstrated that three distinct isoforms of Raldhs of the ALDH1A class exist in vertebrates (ALDH1A1 or Raldh1, ALDH1A2 or Raldh2, and ALDH1A3 or Raldh3) and one Raldh of ALDH8 class called Raldh4. Rodents have an additional ALDH1A enzyme called ALDH1A4 in rat and ALDH1A7 in mouse [28].
RETINOID METABOLISM AND DIABETES-EVIDENCE FROM HUMAN STUDIES
The association of retinoid metabolism and insulin sensitivity, obesity and metabolism has been studied in humans. Basically, the results of these studies are inconsistent and it's difficult to draw a conclusion based on previous results. In the study by Krempf et al. [29], serum vitamin A level was significantly decreased in type 1 diabetes and increased in type 2 diabetes compared with non-diabetic controls. In another study, serum vitamin A level was decreased in young type 1 diabetes [30]. When the vitamin A level was measured in subjects with impaired glucose tolerance (IGT) status, serum vitamin A levels were higher in IGT subjects compared with normal controls [31]. Furthermore, there was a univariate association between vitamin A levels and insulin resistance assessed by the homeostasis model assessment index. Another interesting study by Erikstrup et al. [32] demonstrated that retinol-binding protein (RBP)-to-retinol ratio was elevated in patients with type 2 diabetes. From these results, we see that serum vitamin A levels are high in the hyperinsulinemic status, such as IGT or type 2 diabetes, and low in the insulin-depleted status, such as type 1 diabetes, although more studies are needed to clarify the mechanism.
RXR is crucial in the function of so many other nuclear receptors, since their heterodimeric partner is RXR. RXR is particularly important as it is the heterodimeric partner for PPAR-γ, a terminal regulator of adipogenesis. As RXR is essential for the function of PPAR-γ, the role and expression of RXR in insulin-sensitive tissues might be of interest. In the study by Codner et al. [33], all 3 isoforms of RXR, α, β, and γ were present in human skeletal muscle extracted through percutaneous biopsy of the vastus lateralis muscle. RXR isoform expression is not significantly different between the groups divided by glucose tolerant status and did not show any relationship with the metabolic parameters measured, including insulin, insulin resistance markers or glucose. They concluded that RXR isoforms were not tightly regulated by the factors related to insulin resistance or diabetes.
Several genetic studies were performed under the hypothesis that genetic variation in the components in the retinoid metabolism pathway might affect glucose metabolism or diabetes in humans. Wang et al. [34] screened 10 exons of the RXR-γ genes in the offspring of patients with type 2 diabetes, and identified 14 single nucleotide polymorphisms (SNPs). They analyzed the association of these SNPs with the metabolic parameters and found that none of the variants or haplotypes were associated with glucose in this population. Instead, three of the four variants detected in the mass screening were associated with fasting free fatty acid levels and two variants were associated with triglyceride levels. They also found out that RXR-γ haplotypes were associated with several measures of pancreatic β-cell function. These studies suggest that, although RXR-γ SNPs show some association with pancreatic β-cell function, it is unlikely that this gene is linked to type 2 diabetes.
The Diabetes Genome Anatomy Project (DGAP) was initiated to use a multi-dimensional genomic approach to characterize the relevant set of genes and gene products as well as the secondary changes in gene expression that occur in response to the metabolic abnormalities present in diabetes. In the integrative analysis of the 16 DGAP data sets in multiple tissues, conditions, array types, laboratories, species and study designs, Park et al. [35] revealed that the gene for retinol saturase (RetSat) is a widely shared component of mechanisms involved in insulin resistance and sensitivity. RetSat is an oxidoreductase that catalyzes the reaction of retinol to 13,14-dihydroretinol (dhretinol), and is induced during adipocyte differentiation [36]. RetSat expression is controlled by PPAR-γ through an intronic PPARγ response element (PPRE) in adipocytes. Ablation of RetSat expression in preadipocytes inhibited adipogenesis, whereas ectopic expression of RetSat enhanced differentiation [37]. RetSat-null mice gained weight and the increased adiposity of RetSat-null mice was associated with up-regulation of PPAR-γ, a key transcriptional regulator of adipogenesis [38]. Based on these results, RetSat appears to be a new candidate factor that highlights the importance of the retinol pathway in diabetes, adipogenesis, and insulin resistance.
RXR-PPAR-γ HETERODIMER: TARGETS IN METABOLIC DISEASES
The RXRs play unique and modulatory roles in multiple pathways in the body since they are expressed in various tissues of the body and they form multiple forms of heterodimers with a large number of other nuclear receptors. There are two types of nuclear receptor heterodimers, that is, "permissive" and "non-permissive" heterodimers. Permissive heterodimers (for example, PPARs, LXRs) can be indistinctly activated by ligands of RXR or its partner receptor and are synergistically activated in the presence of both ligands [39]. In non-permissive heterodimers (for example, thyroid receptors, vitamin D receptors), the ligand-induced transcriptional activities of RXR are suppressed. Thus, in these complexes, RXR is a "silent partner." For permissive heterodimers, RXR agonist can demonstrate pharmacological activities that reflect activation of these partners.
Although rexinoids and PPAR-γ agonists produce insulin sensitization in rodent models of type 2 diabetes, the two compounds produce differential tissue-specific gene regulation. Using the Zucker diabetic fatty rat as a model, Singh Ahuja et al. [40] compared the effects of a PPAR-γ agonist, rosiglitazone, and a Rexinoid, the RXR-specific ligand (LG268) on metabolic gene expression in white adipose tissue, skeletal muscle and liver. They showed that rosiglitazone decreased the mRNA expression of tumor necrosis factor-alpha (TNF-α) and increased the mRNA expression for glucose transporter 4 (GLUT4), palmitoyl-transferase (MCPT), stearoyl CoA desaturase (SCD1), and fatty acid translocase (CD36) in white adipose tissue. In contrast, LG268 increased TNF-α mRNA and had no effect, or suppressed the mRNA levels of GLUT4, MCPT, SCD1, and CD36. However, these two compounds showed differential effects in mRNA levels of the genes in liver compared with that of adipose tissue, suggesting the antidiabetic effects of RXR agonists are not just a reflection of RXR-PPAR-γ activation, but may act through complicated pathways.
Treatment of diabetic (db/db) mice with either LG268 or rosiglitazone significantly increased insulin-stimulated glucose transport in skeletal muscle [41]. LG268 increased insulin-stimulated insulin receptor substrate (IRS)-1 tyrosine phosphorylation and Akt phosphorylation in muscle. In contrast, rosiglitazone increased adenylyl cyclase-associated protein gene expression and insulin-stimulated SH3-domain kinase binding protein 1 (SH3KBP1) phosphorylation without affecting the IRS1/Akt pathway. Another study by Li et al. [42] showed similar results when Zucker diabetic rats were treated with AGN194204, an RXR agonist or troglitazone, a PPARγ agonist. These data also suggest that rexinoid and rosiglitazone produce insulin sensitization in skeletal muscle via distinct pathways.
In contrast, studies using antagonists for PPAR-γ and RXR showed different results. In the study by Yamauchi et al. [43], they investigated the phenotype resulting from functional antagonism of PPARγ/RXR, using a PPARγ antagonist and a RXR antagonist in mouse models. Moderately decreased RXR/PPARγ activity in wild-type mice treated with PPARγ antagonist and RXR antagonist showed decreased triglyceride (TG) content in white adipose tissue (WAT), skeletal muscle, and liver. The antagonists also potentiated the effect of leptin and increased fatty acid combustion and energy dissipation, thereby ameliorating-induced obesity and insulin resistance. Paradoxically, when the RXR/PPARγ activity was decreased markedly by treatment of heterozygous PPAR-γ-deficient mice with an RXR antagonist or a PPARγ antagonist, there was depletion of WAT, marked decrease in leptin levels and energy dissipation, and increased TG content in skeletal muscle and liver, thereby leading to the re-emergence of insulin resistance. These data suggest that appropriate functional antagonism of PPARγ/RXR may be a logical approach to protect against obesity and related diseases such as type 2 diabetes.
The most striking differences between rexinoids and PPARγ agonists are their influence on body mass gain and food consumption. The thiazolidinediones (TZDs) are known to produce increased body weight and fat mass. Rexinoids reduce food consumption, body mass gain and fat mass in insulin-resistant or diabetic rodents [44,45]. Another difference between rexinoid and TZDs are the effects on TG levels and lipoprotein lipase (LPL) activity. TZDs have been shown to decrease circulating TG levels and to increase LPL activity. Rexinoids increase TG and decrease LPL activity in heart and skeletal muscle [46].
RETINOID METABOLISM AND INSULIN SECRETION
Pancreatic β-cell dysfunction is one of the most important pathogenic mechanisms in the development of type 2 diabetes, and the most important pathogenic mechanism in type 1 diabetes. Type 2 diabetes develops when the insulin secretory function in pancreatic islets cannot compensate for the hyperinsulinemia that develops due to insulin resistance.
AtRA induces pancreas development and differentiation into acini, but restricting dietary vitamin A in diabetic-prone rats reduces diabetes and insulitis, suggesting the important role of atRA in the development of pancreas islets and in the prevention of pancreatic dysfunction [47-49]. It is evident that RA affects the function of pancreatic β-cells: 1) it restores insulin secretion in vitamin-deficient rats [50], 2) it induces both 1st and 2nd phase insulin secretory responses to glucose in explants of human fetal pancreas [51], 3) it increases insulin production in insulin-secreting cell lines [52], and 4) retinoid acid increases pancreatic glucokinase activity and mRNA levels in the insulinoma cell line and primary rat pancreatic islets [52]. Using the insulin-producing cell line, INS-1, Blumentrath et al. [53] revealed that RAR and RXR were present in INS-1 cells and that 9cRA and atRA inhibited the proliferation of INS-1 cells. They also showed that a moderate concentration (100 nM) of atRA and 9cRA increased glucose-stimulated insulin release in these cells in parallel with GLUT2 expression.
The significant difference that 9cRA has over atRA is that 9cRA is the only ligand for RXR, which serves as an obligatory partner for RAR and numerous other NRs that regulate metabolism and energy balance. Applying a liquid chromatography/tandem mass spectrometry assay, Kane et al. [54] detected not only atRA, but also 9cRA in the mouse pancreas. They showed that 9cRA decreases with feeding and after glucose dosing and varies inversely with serum insulin. 9cRA reduces glucose-stimulated insulin secretion in mouse islets by reducing GLUT2 and glucokinase activity. 9cRA also reduces Pdx-1 and HNF5α mRNA expression. Pancreatic β-cells generate 9cRA, and mouse models of reduced β-cell number or diabetes have reduced 9cRA, in contrast with abnormally high expression of 9cRA in mice with diet-induced obesity and β-cell hyperplasia, suggesting 9cRA is a pancreatic-specific autacoid with multiple mechanisms of action in pancreatic insulin secretion. In contrast, Chertow et al. [55] reported that 9cRA stimulates insulin secretion from the same cell line, possibly through RXR in the same cell line, and they suggested that these effects are greater than that observed with atRA, opposing the results from the previously mentioned group.
A very recent study suggested that a new member in the retinoid metabolic pathway might be involved in pancreatic insulin secretory function. Karasawa et al. [56] reported that the pancreatic islet isolated from HFD-fed BDF1 mice showed a reduced insulin content and glucose-induced insulin secretion from these islets was also significantly impaired with simultaneous increases in glucagon-positive cells, suggesting the expansion of pancreatic α-cells. In the comprehensive gene expression analysis of the pancreatic islets of HFD-fed BDF1 mice and spontaneously diabetic C57BL/KsJdb/db mice, retinaldehyde dehydrogenase 3 (Raldh3) expression was significantly increased [57]. Exposure to higher glucose concentration significantly increased Raldh3 expression in murine β and α cell lines, and overexpression of Raldh3 reduced insulin secretion in β-cells and increased glucagon secretion in α-cell lines. Furthermore, knockdown of Raldh3 expression decreased the glucagon secretion in α-cell line. Given that Raldh3 catalyzes the conversion of 13-cis-retinaldehyde to 13-cis-RA, they also revealed that 13-cis RA significantly reduces cell viability in insulin-secreting cell and α-cell lines. These results suggest that Raldh3 might be a novel factor that regulates the balanced mechanisms of insulin and glucagon secretion in the pancreatic islets.
There are studies showing the effect of RXR activation on pancreatic insulin secretory function [58]. In spontaneously diabetic C57BL/KsJdb/db mouse models, both RXR agonist and PPARγ agonists, when given for 14 weeks, showed significant hypoglycemic effects and the insulin content in islets from both groups showed significant increases compared with controls, suggesting the equal effectiveness of the two agents on increasing pancreatic insulin content.
EFFECTS OF RETINOID ON THE PREVENTION OF TYPE 1 DIABETES THROUGH THE MODULATION OF IMMUNE FUNCTION
Type 1 diabetes is an autoimmune disorder with numerous genetic susceptibility loci, including major histocompatibility complex (MHC) and a much larger list of immunomodulatory environmental factors that have a negative or positive influence on pancreatic β-cells [59]. Vitamin A is considered to be one of the "immunomodulatory" factors that affects the progression of type 1 diabetes, and it is known to be deficient in type 1 diabetes patients [60]. There have been several studies on the beneficial effects of retinoids on the modulation of immune function in the development of type 1 diabetes in animal models.
In the study by Van et al. [61], they used two ways to prove that retinoid affects the development of type 1 diabetes in a specific mouse model. When the splenocytes from newly diabetic non-obese diabetic (NOD) mice were adoptively transferred to NOD/SCID recipient mice, the control recipient mice developed diabetes within 3 weeks. However, atRA treatment not only significantly delayed the onset of diabetes for 5 weeks, but also significantly reduced diabetes incidence compared with controls. Histological studies showed that while pancreas from recipient mice showed severe destructive insulitis, either intact islets or stationary peri-insulitis were observed in atRA-treated mice. These results were due to the fact that atRA treatment induced T regulatory (Treg) cell-dependent immune tolerance by suppressing both CD4+ and CD8+ T effector cells, while promoting Treg cell expansion. These results demonstrate that atRA treatment has immunomodulatory effects on the prevention of type 1 diabetes through the effects on Treg and Teff cells.
Another study examined the effects of the synthetic vitamin A derivative, etretinate and atRA in three preclinical models of type 1 diabetes, spontaneous and cyclophosphamide (CY)-accelerated diabetes in NOD mice, and diabetes induced in a susceptible rodent strain by multiple low doses of streptozotocin (MLD-STZ) [62]. When etretinate and atRA were administered prophylactically to MLD-STZ mice, both drugs effectively prevented clinical signs of diabetes with reduced emergence of autoreactive CD4+ CD25+ T cells, but not with the emergence of Foxp3+ Treg in the peripheral compartment. In spontaneously diabetogenic NOD mice, atRA prophylaxis markedly reduced hyperglycemia and incidence of diabetes. However, in the CY-NOD mice, in which the proportion and function of CD4+ Foxp3+ Treg cells were abrogated with cyclophosphamide, atRA failed to show protective effects on type 1 diabetes, suggesting the that effectiveness of T1DM suppression by retinoids depends on the presence of Tregs, which down-modulate immunoinflammatory events.
These results indicate that atRA exerts a protective effect on type 1 diabetes development through the modulation of immune function, especially through the expansion of Treg cells.
RETINOID METABOLISM AND INSULIN SENSITIVITY
There are several studies relating insulin resistance and the components of retinoid metabolism. The main link is the effect of RXR on glucose metabolism. Treatment of obese and diabetic mice with RXR pan-agonists improves glucose metabolism [63]. However, it is unknown which RXR isoforms are involved in glucose metabolism and which organ is being targeted. Among the three RXR isoforms, RXRγ is preferentially expressed in the skeletal muscle, and its expression is found to change according to nutritional status. Overexpression of RXRγ in mice showed higher glucose disposal than in control mice, and the skeletal muscle from mice overexpressing RXRγ showed increased GLUT1 expression in an insulin-independent manner [64]. Microarray data showed that RXRγ overexpression resulted in the increased expression of a diverse set of genes, including glucose metabolism genes.
Chronic feeding of a vitamin A-rich diet to adult male obese rats for 2 weeks decreased body weight gain and visceral WAT mass [65]. However, vitamin A had no impact on fasting plasma glucose levels/insulin sensitivity. In another study from the same group, 50-day-old young male lean and obese rats were fed with either stock diet or vitamin A-enriched diet for 3 months [66]. Compared with stock diet-fed obese rats, vitamin A-enriched diet-fed obese rats had reduced body weight gain, visceral adiposity and improved insulin sensitivity as evidenced by decreased fasting plasma insulin and unaltered glucose levels, which was due to higher phosphorylation of the soleus muscle insulin receptor. This is explained by decreased protein tyrosine phosphatase 1B levels in the soleus muscle.
In a recent review by Berry and Noy [67], they tried to explain the mechanism for the insulin resistance caused by the increased level of RBP in blood. They suggested that STRA6, a cell surface transporter that binds RBP and facilitates the movement of retinol from the serum protein into cells, is not only a vitamin A transporter, but also functions as a surface signaling receptor. Binding of retinol-bound RBP (RBP-ROH) to STRA6 induces the phosphorylation of a tyrosine residue in the receptor C-terminus, thereby activating a JAK/STAT signaling cascade. Consequently, in STRA6-expressing cells such as adipocytes, RBP-ROH induces the expression of STAT target genes, including SOC3, which suppresses insulin signaling, and PPARγ. Whether RBP-ROH and STRA6 are involved in other biological functions remains to be clarified.
From these results, there is no clear evidence linking retinoid metabolism with insulin sensitivity. More studies are needed to assess the role of retinoid metabolism in insulin sensitivity in various organs involved in insulin signaling.
RETINOID METABOLISM AND HEPATIC GLUCONEOGENESIS
Abnormal hepatic lipid and glucose metabolism significantly contribute to insulin resistance and metabolic derangements. The main key abnormalities are the altered expression of genes involved in lipid and glucose metabolism.
Glucokinase (GK) is an enzyme that phosphorylates glucose to glucose-6-phosphate, which is the first step of both glycogen synthesis and glycolysis. GK is exclusively expressed in hepatocytes, and when the glucose level is high, GK serves as a central metabolic switch to shift hepatic carbohydrate metabolism between fed and fast states [68]. In the study by Chen et al. [69], they made lipophyllic extract (LE) from rat liver and they found that LE synergized with insulin to induce Gck mRNA expression in primary hepatocytes. The active molecule in LE was identified as retinol and Rald by mass spectrometry. Retinoids synergize with insulin to induce Gck expression through the activation of both RAR and RXR. The in vivo study with Zucker lean rats fed with a vitamin A-deficient (VAD) diet demonstrated that hepatic GK activity and Gck mRNA levels were significantly lower than those of rats fed with a vitamin A-sufficient (VAS) diet. These results suggest that retinoids synergize with insulin to induce hepatic Gck expression.
The authors from the above-mentioned study also reported on the synergistic effects of retinoids with insulin on sterol-regulatory element binding protein 1-c (Srebp-1c) expression in primary hepatocytes [70]. Induced expression of Srebp-1c is followed by the elevation of fatty acid synthase (Fas), its target gene. Activation of RXR, but not RAR, is responsible for the induction of Srebp-1c expression. The RA-responsive elements in the Srebp-1c promoter were previously identified as two liver X receptor elements responsible for mediating insulin action. These results indicate a role of vitamin A in the regulation of hepatic gene expression.
RBP4 AND DIABETES
RBP4 is a member of the lipocalin family of proteins that transport small hydrophobic molecules [71]. RBP4 transports retinol from the liver to the peripheral tissues and plasma RBP4 levels positively correlate with retinol levels. In a recent study, the ratio between RBP and retinol was measured in 233 humans divided into three groups depending on normal glucose tolerance (NGT), IGT and type 2 diabetes [32]. The RBP4-to-retinol ratio was higher in type 2 diabetes compared with NGT subjects and was correlated positively with 2-hour postprandial glucose levels, suggesting that RBP-to-retinol is more indicative of type 2 diabetes than RBP itself. When the retinol:RBP4 ratio was decreased by administration of atRA in diabetic ob/ob mice, it improved insulin sensitivity and lowered body weight [72].
The role of RBP4 in insulin resistance and obesity was discovered by Barbara Kahn's group in Beth Israel Deaconess Hospital, Boston, USA. They found that the mice with an adipose-specific knockout of GLUT4 developed insulin resistance and from DNA assays, they found that RBP4 expression was increased in adipose tissue of adipose GLUT4-/- mice and reduced in mice overexpressing adipose GLUT4 [73]. Serum RBP4 levels were increased and positively correlated with body mass index in obese non-diabetic and diabetic subjects [74]. Also, increased RBP4 levels were linked to increased visceral adipose tissue content.
However, the associations between RBP4 levels and measures of obesity or insulin resistance were not consistent. Several studies found no correlation between RBP4 levels and obesity or the amount of adipose tissues [75,76]. Other studies failed to find a significant correlation between RBP4 levels and insulin resistance, IGT, type 2 diabetes or altered insulin secretion. The suggested explanations for these discrepancies are the differences in the renal clearance of RBP4 caused by the different renal functions in the study subjects, the imbalance between RBP4 and retinol, the influence of the collection method, and the antibodies to measure RBP4 in immunoassays [77].
The synthetic retinoid, fenretide (N-(4-hydroxyphenyl)retinamide, FEN) reduces serum RBP4 levels in rodents and humans by disrupting the ternary complex of retinol-RBP4-transthyretin and thereby promoting renal clearance of RBP4. When wild-type mice were fed a HFD with or without FEN, FEN treatment reduced HFD-induced adiposity and hyperleptinemia. FEN improved insulin action on glucose uptake and glycogen levels in muscle, insulin-stimulated suppression of hepatic glucose production and suppression of serum FFA levels in HFD mice [78]. These effects are thought to be independent of the RBP4-lowering effect, since similar effects were observed in RBP4-knockout mice.
In conclusion, RBP4 is an adipokine and a protein that transports retinol to the target tissues. Although there is still debate, RBP4 functions as a link between insulin sensitivity, obesity and type 2 diabetes. RBP4 might also be a clue to the relationship between retinoid metabolism and insulin sensitivity and furthermore, diabetes. More specialized and targeted research has to be performed to uncover the basic role of RBP4 in metabolic diseases.
THE EFFECTS OF RETINOID THERAPY ON INSULIN SENSITIVITY AND LIPID PROFILES IN HUMANS
Although experimental evidence supports the role of retinoid treatment on glucose metabolism and adipogenesis in animals, inconsistent data are available from humans. Most human studies are based on post-treatment data obtained from patients treated with isotretinoin, a 13-cis retinoic acid, for acne, and etretinate, a synthetic retinoid, for psoriasis. Etretinate therapy for psoriasis was reported to be associated with a reduction in glucose levels in response to glucose load [79]. The treatment with acitretin, an aromatic all-trans retinoic acid, showed a mild, transient reduction of insulin sensitivity and high density lipoprotein cholesterol levels, and was not related to any modifications of adipocytokine levels [80].
Isotretinoin is an effective drug for the treatment of acne, but elevated liver enzymes, dyslipidemia, insulin resistance, and type 2 diabetes during isotretinoin therapy have been reported [81]. The mechanism underlying hypertriglyceridemia in retinoid-treatment is thought to involve impaired clearance of triglyceride-rich particles. In the study by Stoll et al. [82], whole body and adipose tissue insulin sensitivity in 15 healthy male volunteers before and after a 5-day administration of isotretinoin was assessed. Isotretinoin treatment increased plasma triglycerides, but did not change whole body insulin-mediated glucose disposal and lipolysis. The authors concluded that impaired clearance of TG-rich particles due to isotretinoin treatment for 5 days does not impair insulin-mediated inhibition of lipolysis or glucose disposal. In a recent report by Ertugrul et al. [83], a total of 48 patients with acne vulgaris were treated with isotretinoin for 3 months. Although liver enzymes, total cholesterol, low density lipoprotein cholesterol and TG levels were elevated compared with baseline after 3 months of treatment with isotretinoin, there was not a significant change in fasting glucose, insulin, C-peptide levels or homeostasis model assessment of insulin resistance (HOMA-IR) values before or after treatment, indicating that there is no effect of isotretinoin treatment on insulin sensitivity. In another study performed in 23 healthy acne patients, 3 months of treatment with 13-cis retinoid acid resulted in increased HbA1c, TG, C-peptide and increased adiponectin levels, suggesting controversial effects of retinoid treatment on glucose metabolism and adiponectin concentration [84].
The results from the previously performed studies on the effects of retinoid treatment on insulin resistance and glucose metabolism did not show consistent results. Although they show some consistent trends, more studies are needed to address their conclusive effects, since most of the studies were not initiated with glucose metabolism as the end point. Therefore, randomized controlled studies with diabetes development or glycemic progression as the end points should be dedesigned and performed to draw any conclusion regarding the effect of retinoid treatment on insulin sensitivity and metabolism in humans.
RETINOID METABOLISM AND LIPID METABOLISM
Vitamin A deficiency is known to favor fat deposition. Extensive evidence has established that RAR and RXR participate in determining adipogenesis. For example, during adipogenesis, RXR expression patterns vary considerably, and the effects of atRA versus 9-cis-RA also vary depending on when cells are stimulated [85]. RAR plays an equal if not more dominant role in adipogenesis [86]. In RAR overexpressing cells, RA inhibits differentiation of 3T3-L1 cells during early stages of adipogenesis, but not when added 48 hours after differentiation has been initiated. Thus, although it is apparent that RA blocks adipocyte differentiation in a certain stage of differentiation, the effect could vary according to the stage, including the levels of transcriptional factors.
Rald has been primarily considered merely as a precursor for RA formation through irreversible oxidation by Raldh [87]. However, recent work suggests that Rald is present in fat tissue where it may exert effects independent of its conversion to RA [88]. Rald levels vary inversely with adiposity in mice fed a HFD, while levels of ADH and Raldh1 are also differentially regulated. Mice lacking Raldh1 are protected against diet-induced obesity and diabetes. Consistent with the notion that Rald acts independently of its conversion to RA, administration of retinol, Rald, or RA to ob/ob mice had divergent effects on adiposity, with Rald limiting increases in visceral adiposity. Interestingly, in toxicology studies, citral, an inhibitor of Raldh activity, also induced weight loss [89]. Rald appears to inhibit RXR:PPARγ activation.
Another novel mechanism relating RA and fat metabolism is the recent observation that RA may have unique effects on lipid metabolism through differential effects on activating RARs versus PPARβ/δ [90]. RA is a proposed ligand for PPARβ/δ, a receptor involved in energy balance, lipid metabolism, and glucose homeostasis. PPARβ/δ activation increases lipid catabolism in skeletal muscle and adipose tissue, preventing the development of obesity [91]. In these studies, adipogenesis was accompanied by altered RA signaling in mature adipocytes, with activation of RARs and PPARβ/δ, thus enhancing lipolysis and depleting lipid storage. In diet-induced obesity, RA treatment induced the expression of RAR and PPARβ/δ target genes involved in regulation of lipid homeostasis, fostering weight loss. Despite these provocative results, more direct studies are needed to understand how retinoids and retinoid-activated receptors modulate lipid metabolism and adipogenesis and affect diabetes.
CONCLUSIONS
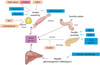
Retinoids might seem a little bit unfamiliar to the readers of a diabetes metabolism journal. However, there are many things to be uncovered regarding the role of retinoids on diabetes development, insulin resistance, pancreatic dysfunction and lipid metabolism (Fig. 3). In general, two components in retinoic acid metabolism, RXR and atRA, are the main components that involve in glucose metabolism. Rexinoids are important in glucose and lipid metabolism, since they heterodimerize with PPARs, which are critical determinants in lipid and glucose metabolism. Although rexinoids are known to function as insulin sensitizers, most of their effects are mediated through the dimerization with PPARγ. While RXR agonist influences primarily insulin sensitivity, retinoid acid, mostly atRA, affects insulin secretion and islet function through immunomodulation. For the rexinoids to be considered as a novel therapeutic target for obesity or diabetes, higher specificity for certain RXR subtypes is mandatory. In addition, an interesting aspect of retinoids is the true function of Rald in metabolic diseases. It is not even clear yet whether Rald is a unique substrate that could function alone as a ligand to the nuclear receptors or just as an intermediate metabolite of retinoid metabolic pathways. There is a lot to learn about the effects and the function of Rald on metabolism. Further, there should be studies performed regarding the possible association between retinoids and the incretin system. If any effects of retinoid on the incretin system are found, retinoid could definitely be considered a new highlighted strategy that provides hope for a diabetes cure.
 XML Download
XML Download