

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The number of people with type 2 diabetes has rapidly and remarkably increased throughout Asia, and the rate of increase shows no signs of slowing. This recent epidemic of type 2 diabetes in Asia differs from that reported in other regions of the world; it has evolved over a much shorter time, at an earlier stage of life, and in people with lower body mass indices [1]. Because the degree of obesity, the aging process and environmental influences on the body are closely related to insulin resistance, insulin secretory defects may perform a more important function in the development and progression of diabetes in our region [2]. Impaired insulin secretion might be induced by insufficient β-cell mass, by functional defects within the β-cells themselves, or by both of these conditions. Reductions in β-cell mass and abnormalities of β-cell function can both be demonstrated in patients with type 2 diabetes and individuals at increased risk for diabetes [3]. Whether β-cell dysfunction is caused by reduced β-cell mass or vice-versa remains to be determined. A genetic element clearly underlies β-cell dysfunction and insufficient β-cell mass; however, a number of modifiable factors are also linked to β-cell deterioration, most notably chronic hyperglycemia and elevated free fatty acid (FFA) levels [4]. Evidence has also been found for a link between increased pro-inflammatory cytokines and the impairment of insulin-signaling pathways in the β-cells, as well as the potential roles of islet amyloid deposition and fibrotic islet destruction [5,6]. In this review, we provide an overview of the characteristic features and underlying mechanisms of dysfunctions and death of the β-cells by glucolipotoxicity.
GLUCO-, LIPO- AND GLUCOLIPOTOXICITY
Glucotoxicity
In type 2 diabetes, chronic hyperglycemia has long been felt to have negative consequences on β-cell function. The results of several studies have demonstrated that the chronic elevation of blood glucose concentration impairs β-cell function and insulin sensitivity, a phenomenon referred to as glucotoxicity [7,8]. Glucose has a stimulatory effect on transcription of the gene that encodes preproinsulin and also on insulin release. Glucose enters the β-cell via facilitated transport through the glucose transporter 2 (GLUT2) transporter, and then it is converted to glucose-6-phosphate (G-6-P) by the action of glucokinase (GK). The glycolytic cascade and adenosine triphosphate (ATP) production in this process ultimately leads to membrane depolarization and exocytosis of insulin granule [9]. It is understandable that repeated and prolonged exposure to hyperglycemia likely leads to β-cell degranulation and eventual exhaustion, but the mechanisms underlying this process are believed to be complex and not readily explicable. Prolonged hyperglycemia alters various β-cell-specific genes, including the transcription factors Pdx-1, BETA2/NeuroD and MafA, and other genes as well, such as insulin, PGC-1α, UCP, and many genes related with the glycolytic pathway [10-13]. The reduction of insulin-gene transcription is thought to be secondary to reductions in the transcription or activity of β-cell transcription factors such as Pdx1, BETA2/NeuroD and MafA [10-14]. Several mechanisms have been proposed to explain the hyperglycemia-induced loss of β-cell function, but a major factor appears to be alteration of intracellular energy metabolism and oxidative stress, as well as mitochondrial dysfunction, which will be discussed later. Other pathways linked to hyperglycemia include endoplasmic reticulum (ER) stress and possibly hypoxia-induced stress [15,16]. Therefore, glucose toxicity is one of the most important mechanisms of β-cell dysfunction and loss in diabetic patients. However, it is difficult to explain β-cell loss observed in the prediabetic stage [17].
Lipid toxicity
The term "lipotoxicity" is often applied to the phenomenon in which elevated FFA levels in the setting of insulin resistance contribute to β-cell dysfunction. In actuality, the effect of FFA on β-cell function is much more complex, and it includes both beneficial and detrimental effects [18]. Duration of exposure to elevated FFA and the coexistence of hyperglycemia influence the extent to which FFA levels contribute to β-cell function. Some FFAs and lipoproteins may exert a pro-apoptotic effect, whereas others appear to perform a protective function. Therefore, long-term exposure to saturated fatty acids (such as palmitate) is associated with toxic effects, whereas monounsaturated fatty acids (such as oleate) protect against palmitate- and glucose-induced β-cell apoptosis [19]. Exposure to FFA in the long-term leads to impaired insulin gene transcription, impaired glucose-stimulated insulin secretion (GSIS) and eventual β-cell apoptosis [20]. Researchers have studied the mechanisms by which healthy concentrations of FFA promote GSIS, and at least two distinct pathways have emerged. The first is through the free fatty acid receptor (Gpr40) [21]. Alquier et al. [22] recently showed that the knockout of GPR40 led to impairments in glucose- and FFA-stimulated insulin secretion in islets without affecting intra-islet glucose or palmitate metabolism. The second pathway is through intracellular FFA metabolism and glycerolipid/FFA cycling [23]. In the aggregate, these mechanisms are believed to maintain GSIS under normal circumstances and possibly contribute to the early hypersecretion of insulin in the initial stages of high-fat diet-induced obesity [24]. Emerging data additionally implicate a possible role for cholesterol metabolism in β-cell lipotoxicity. Oxidized low density lipoprotein particles appear to diminish insulin gene transcription and promote apoptosis in isolated β-cells [25]. Disruption of the ABCA1 reverse cholesterol transporter in mice results in defects in cholesterol efflux from the β-cell and subsequent accumulation of intra-islet cholesterol; this accumulation leads to impaired GSIS and glucose intolerance [26]. In this regard, recent studies by our group suggest that activation of ABCA1 in human islets by LXR agonists might be one approach to diminish islet cholesterol burden and to improve GSIS [27].
Glucolipotoxicity
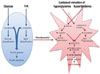
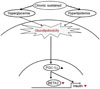
This finding is consistent with the glucolipotoxicity hypothesis, which states that neither glucose nor FFAs alone cause clinically meaningful β-cell toxicity [3]. The term "glucolipotoxicity" is perhaps more appropriate in describing the phenomenon. The effect of glucose on FFA toxicity in the β-cell has been suggested to be secondary to a partitioning effect on lipid metabolism [28]. Several mechanisms have been proposed to explain the chronic effects of FFA on GSIS and β-cell apoptosis. Prolonged exposure to palmitic acid diminishes insulin gene transcription and GSIS in isolated rat islets, and it is accompanied by attenuated binding of the β-cell-specific transcription factors Pdx1 and MafA [29]. Other mechanisms of glucolipotoxicity include palmitic acid-induced synthesis of ceramides, which inhibits the anti-apoptotic protein Bcl-2 and downregulates IRS-1/2 signaling [19,30]; FFA-induced up regulation of UCP2 and reduction of glucose-stimulated ATP generation [31,32]; and activation of the oxidative stress [33] and the unfolded protein response [34] pathways. Elevated glucose concentration alone is not toxic to islet tissue in normal or prediabetic stages. The β-cell adapts via changes in gene expression, such as glycolytic and anaplerotic genes induction [35,36]. These results induce GSIS and glucose detoxification in the pancreatic β-cell [37]. Elevated fatty acids alone also are nontoxic because FFAs will be oxidized in the β-cell at low glucose and in the absence of increased malonyl-CoA levels, resulting in lipid detoxification [38]. However, prolonged exposure to high glucose and FFAs would subsequently result from the progressive accumulation of FFA-derived long-chain acyl-CoA esters (FACoAs) and various lipid-signaling molecules in the β-cell. These factors may cause impaired glucose-induced insulin secretion and biosynthesis and promote apoptotic cell death (Fig. 1). Recently, our laboratory also demonstrated the depression of important pancreas key transcription factors by glucolipotoxicity. Kim et al. [13] reported that PGC-1α inhibits insulin and BETA2/NeuroD transcription levels and that attenuating PGC-1α overexpression protects against glucolipotoxicity-induced β-cell dysfunction. Although the basal PGC-1α expression level was very low in fresh, isolated rat islets in our study, it increased gradually with time under glucolipotoxic conditions. After three days of exposure to such high glucose concentrations, insulin and BETA2/NeuroD gene expression levels were downregulated significantly. An even more interesting finding was that the suppression of the Pdx-1 gene needed more time after exposure to glucolipotoxic conditions, and then it gradually decreased. We suggest that PGC-1α played important key roles in intracellular fuel regulation and could herald in a new era in the treatment of patients with type 2 diabetes mellitus by providing protection from glucolipotoxicity, which is an important cause of the development and progression of the disease (Fig. 2).
STAGES OF GLUCOLIPOTOXICITY
We viewed the changes that occurred in β-cells during the progression from the normal state to severe diabetes as consisting of three stages (Fig. 3). We cited much of stages of glucolipotoxicity in a similar manner as those referenced in Weir et al. [39].
Stage 1: mild decompensation
In this stage, there is a specific loss of GSIS, whereas insulin secretion to other secretagogues, such as arginin, IBMX and GLP-1, is preserved. Insulin stores in the β-cells are well preserved, suggesting that secretory mechanisms (including channel activities, membrane docking of insulin granules and mitochondrial ATP production) are more severely affected than those of insulin synthesis [40]. A loss of β-cell differentiation can be found at this stage, but insulin mRNA levels are protected, which probably allows insulin production to be reasonably well maintained for the degree of β-cell deficiency.
Stage 2: severe decompensation
β-cells are degranulated, which coincides with a decrease in insulin mRNA, pointing to decreased insulin synthesis. In this situation, β-cell differentiation is grossly deranged, with alterations of metabolic genes and key transcription factors (Pdx-1, BETA2/NeuroD, and MafA), as well as increased expression of several important stress genes (A20 and Heme oxygenase-1).
Stage 3: decompensation with structural damage and death of β-cells
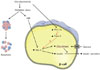
Islets can transition from having a relatively normal structure with abnormal function to a stage of obvious structural damage. Amyloid formation in human type 2 diabetes is a striking abnormality. Amyloid fibrils can have a destructive effect on β-cells, but we know little about the mechanisms responsible for their formation. Lipid droplets are sometimes found in β-cells, which raises questions about lipotoxicity. Islet fibrosis can also be found. Although it is tempting to suggest that an increased rate of apoptosis must be important for the development of human type 2 diabetes, it is possible that limitations of islet neogenesis and β-cell replication are at least as important. Severe β-cell ER stress leading to strong activation of the UPR might cause apoptosis, which is mediated by stress kinases and transcription factors, such as Jun N-terminal kinase (JNK) and C/Ebp-homologous protein (CHOP). Importantly, disruption of the CHOP gene was shown to inhibit β-cell apoptosis, expand β-cell mass and improve glycemic control in mouse models of diabetes, which suggests that the UPR plays an important role in mediating the β-cell dysfunction of diabetes. We found that glucose moderately stimulates ER stress; however, high glucose levels synergize with fatty acids to stimulate UPR and JNK with β-cell apoptosis as a consequence. Chronic exposure to high glucose is expected to increase the metabolic flux in mitochondria and lead to excess production of reactive oxygen species (ROS) through the hexosamine pathway. The level of oxidative stress exerted on the β-cell depends on its capacity to scavenge ROS and other free radicals generated under conditions of glucolipotoxicity. Oxidative stress and/or activation of the JNK pathway suppresses insulin gene expression, which is accompanied by reduction of PDX-1 [41]. Interestingly, elevation of PGC-1α by glucolipotoxicity might promote or enhance the harmful effects of the JNK pathway (Fig. 4).
SUMMARY AND CONCLUSION
Progressive loss of β-cell function and mass with aging are observed in type 2 diabetic patients and even in people with normal glucose tolerance. A genetic element clearly underlies β-cell dysfunction and insufficient β-cell mass; however, a number of modifiable factors are also linked to β-cell deterioration, most notably chronic hyperglycemia and elevated FFA levels. Pancreatic β-cells exposed to high glucose and lipid concentrations display altered gene expression, function, survival, and growth that may contribute to the slow deterioration of the functional β-cell mass in type 2 diabetes. These glucolipotoxic alterations may result from various types of stress, including oxidative stress, ER stress, cytokine-induced apoptosis, and hypoxia. A clearer understanding about the differences of energy metabolism between normal and pathologic conditions and related alterations of important genes expression in β-cells should help explain the mechanism of glucolipotoxicity in β-cells.

 XML Download
XML Download