

 PDF
PDF ePub
ePub Citation
Citation Print
Print


PEROXISOME PROLIFERATOR-ACTIVATED RECEPTORS
Peroxisome proliferator-activated receptors (PPAR) are ligand-activated transcription factors that are members of the nuclear hormone receptor superfamily [1,2]. There are three PPAR isoforms of the distinct genes commonly designated as PPARα (NR1C1), PPARγ (NR1C3) and PPARβ/δ (NR1C2) (or simply δ) [1]. The identification and designation of this PPAR subfamily of nuclear receptors in the 1990s was the result of over 25 cumulative years of work with peroxisome proliferators [2]. The PPARs heterodimerize with another nuclear receptor, the 9-cis-retinoic acid receptor (RXR), to form a complex that interacts with specific DNA-response elements within the promoter regions of the target genes. This heterodimer complex is activated by appropriate ligand binding, so it can therefore recruit transcription coactivators and oversee the transcription of genes involved in the regulation of lipid and carbohydrate metabolism [3].
Tissue expressions differ based on the PPAR subtype [4,5]. PPARα is highly expressed in the liver, renal cortex, intestinal mucosa, and heart, which are all organs that possess high mitochondrial and β-oxidation activity. Lower expression of PPARα is also observed in several other tissues. Similar tissue expression profiles of PPARα have been found in rodents and humans [4]. PPARα is abundantly expressed in the proximal tubules and the medullary thick ascending limbs, and to a lesser extent, in the glomerular mesangial cells [6,7]. Given the high level of expression in the renal proximal tubules, PPARα has been implicated in the metabolic control of the kidney in maintaining a sustained balance of energy production and expenditure.
PPARs basically function as sensors for fatty acid derivatives and control important metabolic pathways involved in lipid and energy metabolism. PPARs also play an important role in various pathophysiologic conditions, such as immunity, inflammation, apoptosis, and cell differentiation [8]. Each member of the PPAR subfamily has additional actions [9,10]. PPARα plays an additional role in lipoprotein synthesis, inflammatory responses and the fatty acid oxidation system [2]. In general, PPARα functions as catabolic regulators of energy [2,11].
Numerous studies in experimental and clinical models have shown the beneficial effects of PPARs in improving organ function in some diseases [12,13]. The PPARα agonists, such as fenofibrate and clofibrate, are traditionally proven lipid-lowering drugs [13]. Despite their serendipitous discovery and clinical use in the treatment of dyslipidemia, the actual pharmacological profile of activity of the PPARα agonists is a new discovery [14]. In addition to their involvement in lipid and lipoprotein metabolism, recent evidence supports the theory that PPARα critically regulates inflammation and vascular function in the kidney, which has led to renewed interest in PPARα as a renal protective option.
ROLE OF PPARα IN NORMAL PHYSIOLOGY
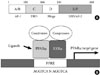
The PPARα gene in the human, which spans ~93.2 kb, is located on chromosome 22q12-q13.1 and, encodes a protein of 468 amino acids. The PPARα gene that encodes mRNA is derived from 8 exons with a 5' untranslated region encoded by exons 1, 2, and part of exon 3 [15]. The remainder of exon 3 and exons 4-8 are known to contribute to the coding region of PPARα. The last 232 bp of exon 8 contribute to the 3'-untranslated region. In the mouse, the PPARα gene is located on chromosome 15E2, and it encodes a protein of 468 amino acids [2]. The encoded protein shares functional domains with other nuclear hormone receptors. As with other nuclear receptors and PPARs, PPARα contains four major functional domains, including the N-terminal ligand-independent transactivation domain (A/B domain), the DNA binding domain (DBD or C domain), the co-factor docking domain (D domain), and the C-terminal E/F domain (including the ligand binding domain [LBD] and the ligand-dependent transactivation domain (AF-2 domain)) (Fig. 1) [13]. The A/B domain contains an activation function-1 (AF-1) region, which has a low level of basal transactivation activity and functions independently of ligand-binding. In humans, DBD encompasses amino acids 101-166, which contain two very highly conserved zinc finger motifs and architectural elements that are capable of sequence-specific binding to DNA [15]. D domain, a flexible hinge domain, connects the DBD and LBD. This hinge region binds co-repressor proteins, with the characteristic LXXXIXXXL repressor motif, to the receptor in its quiescent, unliganded state [2]. LBD in the human PPARα protein, which contains an AF-2 region composed of two α-helices flanking one four-stranded β-sheet, extends from amino acids 280 to 468 [15]. The AF-2 domain is repressed until ligand-binding occurs. Following ligand-binding, the AF-2 domain undergoes a conformational shift, which allows the formation of hydrogen bonds between Tyr-314 and Tyr-464 as well as the formation of a charge clamp between Glu-462 and Lys-292. This conformational change in the protein allows interaction of the receptor with the LXXLL (L, leucine; X, any amino acid) motifs located in co-activator proteins [16]. In a similar fashion as other nuclear receptors, PPARα undergoes conformational adjustment upon binding to a ligand to achieve the co-regulator exchanges and activation of the target genes [2,16].
PPARα, like the other two PPAR isoforms, is localized to the nucleus, which is characteristic of the type II nuclear receptor family [2]. PPARα regulates many target genes, and the expression of the PPARα gene is also affected by other transcription factors [17]. According to an experimental study, PPARα is regulated by various physiological conditions such as stress, hormones, glucocorticoids, insulin, and leptin [18]. Its expression additionally appears to be related to aging [19]. PPARα is also regulated at the transcriptional level by nuclear receptors such as hepatocyte nuclear factor 4 (HNF4) and the orphan receptor, known as chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII). HNF4 positively affects PPARα expression via a direct repeat 1 (DR1) element, which is composed of the consensus sequence AGG(A/T)CA separated with a single nucleotide spacing between two repeats [17]. The DR1 element in the human PPARα promoter is antagonized by COUP-TFII. PPARα also appears to modulate its own expression [20]; transcript levels are induced during macrophage differentiation by high glucose levels, and PPARα is regulated by the ubiquitin proteasomal degradation system [21,22].
PPARα functions as an obligate heterodimer with another nuclear receptor: retinoid X receptor (RXR; NR2B) [2]. The PPARα/RXR heterodimers bind to a specific DNA sequence element called a peroxisome proliferator response element (PPRE), which is located in the promoter region of the target genes [23,24]. PPRE consists of a direct repeat of hexametric core recognition elements spaced by 1 bp (DR1, 5'AGGTCANAGGTCA-3') located in the promoter regions of target genes [13]. The upstream extended hexamer of DR1 interacts with PPARs, whereas the downstream hexamer is known to interact with RXR of the PPAR/RXR heterodimer [15]. Interestingly, PPARγ binds more strongly than the other isoforms, while conservation of the 5'-flanking extension is more important for the binding of PPARα and PPARβ/δ [25]. After activation of the PPARα/RXR heterodimer at the PPRE, the PPARα/RXR complex recruits diverse nuclear receptor co-factors that modulate transcriptional activity of the PPAR and RXR receptor heterodimer [13]. As with other nuclear receptors, transcriptional activation of PPAR genes involves the participation of many transcription co-regulators, and PPARs interact with co-activators, such as steroid receptor co-activator-1 (SRC-1), or co-repressors, such as the nuclear co-repressor (N-CoR) and the silencing mediator for retinoid and thyroid hormone receptors (SMRT) [26,27]. PPARα-interacting co-activators and co-repressors augment or repress, respectively, the PPARα transactivation activity [28].
PPAR activities are regulated by postranslational modification such as phosphorylation. This phosphorylation of PPARα is mediated by insulin and stress. Stress stimuli cause an increase in PPARα phosphorylation in rat neonatal cardiac myocytes via the p38 mitogen-activated protein kinase (MAPK) pathway [29]. PPARα is also reported to be phosphorylated by protein kinase C (PKC). Inhibition of PKC activity impairs ligand-activated PPARα transactivation activity but enhances PPARα transexpression activity, which suggests that the PKC signaling pathway may act as a molecular switch for the transactivation and transexpression properties of PPARα and also that PKC phosphorylation may play a role in statin-mediated anti-inflammatory effects [30,31].
PPARα basically regulates all three fatty acid oxidation systems, mitochondrial and peroxisomal β-oxidative processing and microsomal ω-oxidation, which indicates that PPARα functions mostly as a catabolic regulator of energy expenditure [32]. In this regard, activation of PPARα by pharmacological intervention proved useful in combating diet-induced obesity-associated complications [33,34]. PPARα also has hypolipidemic effects, which shows that PPARα ligands reduce VLDL production and enhance the catabolism of triglyceride (TG)-rich particles. This process indirectly decreases small dense LDL particles, enhancing the formation of HDL particles and hepatic elimination of excess cholesterol [35].
PPARα IN PATHOLOGICAL CONDITIONS
In association with their critical role as a primary sensor and regulator of lipid metabolism, PPARα agonists have been reported to decrease inflammation. According to numerous experimental studies, PPARα appears to influence both acute and chronic inflammatory disorders involving neutrophils and macrophages. Since some preference for specific fatty acids by each PPAR has been demonstrated, fatty acids and their derivatives (including 8(S)-hydroxyeicosatetraenoic acid, lipoxygenase metabolite leukotriene B4 [LTB4], and the arachidonate mono-oxygenase metabolite epoxyeicosatrienoic acids) have been shown to activate PPARα [13]. Among them, LTB4 is a powerful chemotactic inflammatory eicosanoid that induces transcription of genes of the β- and ω-oxidation pathways that neutralize and degrade LTB4 itself to regulate the inflammatory response [2,35]. Without PPARα regulation, the LTB4-induced inflammatory response tends to continue. Furthermore, experimental agents containing LTB4 (or its precursor arachidonic acid) when applied to the ears of PPARα knockout and wild-type mice showed that the inflammatory response was significantly prolonged in PPARα-null mice compared to the wild-type controls, suggesting that PPARα affects the duration of the inflammatory response, possibly by limiting cytokine expression and also by inducing genes that metabolize LTB4 [35]. In addition, PPARα activation may result in reduced leukocyte adhesion to activated endothelial cells of the arterial lumen and subsequent inhibition of the formation of macrophage foam cells by regulating the expression of genes involved in reverse cholesterol transport and reactive oxygen species (ROS) output [36]. PPARα agonists seem to inhibit lipopolysaccharide activation of peritoneal macrophages, indicating a role in inflammation that is independent of macrophage polarization [37]. Fibrate treatment also has been reported to reduce atherosclerosis in apoE-deficient mice and in human ApoAI transgenic apoE-deficient mice [38]. Therefore, activation of PPARα may be beneficial in ameliorating the formation and progression of atherosclerotic plaques by minimizing lipoprotein oxidative modifications.
According to several experimental studies, PPARα ligands influence the levels of pro-inflammatory cytokines, such as interleukin (IL)-1, IL-6, tumor necrosis factor-α (TNF-α), cyclooxygenase-2, and inducible nitric oxide synthase (iNOS). They regulate these cytokines by inhibiting the translocation of the p65 subunit of nuclear factor κB (NF-κB), increasing IκB (the inhibitor of NF-κB) and decreasing phosphorylation of the c-jun subunit of AP-1 [37,39]. PPARα agonists appear to inhibit TNF-α-induced vascular cell adhesion molecule (VCAM)-1 expression in endothelial cells by suppressing transcriptional activity of NF-κB [37]. Considering that PPARα is expressed in both vascular endothelial cells and smooth muscle cells, PPARα can therefore be expected to be involved in vascular pathologic processes. PPARα activation has been reported to inhibit vascular smooth muscle cell (VSMC) proliferation by suppressing telomerase activity through the p16/retinoblastoma/E2F transcriptional pathway. In addition, PPARα agonists have been shown to inhibit inflammation in VSMCs; conversely, PPARα deficiency leads to more profound inflammation in these cells [37]. PPARα has the ability to inhibit vascular inflammation, oxidative stress, and cell growth and migration, as evidenced by experimental results that showed that PPARα blocked NF-κB, transforming growth factor (TGF)-β/Smad and MAPK pathways [40]. Additionally, several other studies have demonstrated a similar, important role for PPARα in modulating inflammation in vascular endothelial cells, cartilage and bone tissue, kidney, adipose tissue, and the central nervous system [2].
PPARα has also been implicated in blood pressure regulation. Experimental studies demonstrated that PPARα agonists reduced angiotensin II-induced hypertension in rats, probably by improving endothelial cell function [41]. A PPARα agonist has also been reported to decrease blood pressure in a deoxycorticosterone acetate (DOCA)-salt-induced hypertensive mouse model by increasing renal 20-hydroxyeicosatetraenoic acid production and therefore decreasing sodium retention [37]. In addition, PPARα has been found to co-localize with arachidonic CYP450 4A enzymes in the renal proximal tubule; knockout cyp4A14 mice exhibited androgen-dependent hypertension [42], which suggested that regulation of CYP4A by PPARα may play a role in sodium homeostasis and blood pressure regulation. Interestingly, PPARα also appears to be associated with tissue renin-angiotensin system (RAS) activity. Our recent study showed that PPARα content in the kidney was negatively correlated with activation of intrarenal RAS [33]. Fenofibrate treatment in spontaneously hypertensive rats fed with a high-fat diet attenuated weight gain, fat mass and insulin resistance, whereas angiotensin receptor blockers or antioxidant treatment did not improve metabolic parameters [33,43]. Besides the recovery of the diet-induced decrease in intrarenal PPARα expression and the increase in lipid accumulation with administration of PPARα agonists, the most important finding was that PPARα abolished intrarenal RAS activation and oxidative stress while also providing protection against increased blood pressure and renal injury [33].
Based on these observations, we surmised that PPARα ligands exerted potential organ-protective effects in modulating inflammatory processes, atherosclerosis, blood pressure, and the RAS.
PPARα AGONIST IN DIABETIC NEPHROPATHY
Hyperglycemia, endothelial dysfunction, lipotoxicity, and dyslipidemia are common denominators of the pathological mechanism that gives rise to diabetic renal complications in both type 1 and type 2 diabetes mellitus [44]. Increasing evidence suggests that PPARα may play critical regulatory roles in a variety of biologic events, such as lipid metabolism, energy homeostasis, insulin sensitivity, inflammation and blood pressure regulation; a therapeutic potential for PPARα agonists in renal complications of diabetes has also been suggested. PPARα has been implicated in the pathogenesis of obesity and insulin resistance, which are among the diagnostic criteria for the metabolic syndrome seen in up to 75% of patients with type 2 diabetes [13].
In vivo experiments
Activation of PPARα reduces weight gain in animals, and a high-fat diet in PPARα-null mice leads to a more dramatic increase in body weight [33,45]. Treatment with PPARα agonists also improves insulin resistance and glycemic control in db/db mice and OLETF rats and prevents the development of diabetes in obese OLETF rats [46,47].
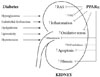
In addition to systemic effects, multiple intrarenal mechanisms have been implicated in the beneficial effect of PPARα ligands (Fig. 2). An experimental study with a type 2 diabetic model showed that PPARα activation by fenofibrate improved insulin sensitivity, glucose control, and diabetic nephropathy, as evidenced by urinary albumin excretion and attenuated glomerular mesangial expansion [48]. Such beneficial effects on renal outcome with fibrates may result from anti-inflammatory, anti-atherosclerotic, antihypertensive, and anti-RAS actions as noted above. In an experimental study to investigate the role of PPARα in type 1 diabetic nephropathy, more severe structural changes (such as glomerulosclerosis and mesangial area expansion) as well as an effect on albuminuria were noted in diabetic PPARα-knockout mice; these changes were associated with an increase in the profibrotic, pro-inflammatory and pro-apoptotic pathways implicated in renal extracellular matrix accumulation [49]. In PPARα deficiency, the glomerular lesions exhibited increased type IV collagen and TGF-β expression in diabetic kidney disease, suggesting that the activation of PPARα ligands effectively prevents the glomerular matrix expansion that accompanies apoptosis and inflammatory cell infiltration in the glomerulus [49].
In our study, which investigated the effect of the glucagon-like peptide-1 analog exendin-4 on the progression of type 2 diabetic nephropathy, we observed significantly increased PPARα expression in a dose-dependent manner in exendin-4-treated db/db kidneys in mice compared with that seen in control db/db kidneys [50]. This increase in PPARα expression was accompanied by reduced glomerular immunostaining for F4/80 and caspase-3 as well as for TGF-β1. Furthermore, exendin-4 treatment decreased 24-hour urinary 8-hydroxy-deoxyguanosine concentration, which was consistent with the reduction in oxidative DNA damage and oxidative stress. These findings suggest that TGF-β1 expression mediated by oxidative stress may be suppressible by PPARα activation [50]. Interestingly, there is evidence that starved PPARα-null mice develop increased albuminuria and exhibit albumin accumulation in the proximal tubules, which indicates that PPARα activity may facilitate albumin reabsorption and degradation in this nephron segment [51]. This mechanism may contribute to the beneficial effect of PPARα agonists on albuminuria in type 2 diabetic nephropathy.
In vitro experiments
In mesangial cells, PPARα agonists reduce the production of TGF-β and extracellular matrix. The TGF-β signaling pathway may be one possible mechanism that relates to the effect of PPARα agonists on the mesangial matrix production. One study showed that clofibrate directly inhibits oxidant stress-induced TGF-β1 expression in these cells, indicating that PPARα agonists block the TGF-β signaling pathway, thereby attenuating glomerular matrix production [52]. PPRE3X luciferase reporter analysis demonstrated that the fenofibrate significantly increased luciferase activity in mesangial cells, which is consistent with the existence of endogenous PPARα activity in these cells [48,49]. This finding suggests that increased PPARα activity in the tubule may exert anti-inflammatory and anti-fibrotic effects via paracrine action resulting from increased PPRE activity in the glomeruli [49].
Importantly, we need to pay attention to the role PPARα in vascular biology. PPARα ligands appear to modulate renal endothelial cell proliferation and migration, probably through their ability to interfere with the vascular endothelial growth factor (VEGF)-mediated signaling pathway. VEGF is crucial for maintaining the function and integrity of the endothelium [44]. In the kidney, the VEGF receptors (VEGFR) are expressed in the endothelium of the glomeruli, the peritubular capillaries and, to a lesser extent, the mesangial and tubular cells [53]. In the glomerular endothelial cells, VEGF-A stimulates the VEGFR-2/Akt axis to regulate endothelial NOS (eNOS) phosphorylation. eNOS is activated by the phosphorylation of serine (Ser1177) of the protein kinase Akt/PKB and is also known to regulate glomerular hemodynamics by generation of nitric oxide (NO) [53,54]. Several type 1 or type 2 animal models have shown that VEGF stimulates renal pathological progressions, as demonstrated by glomerular hyperfiltration and hypertrophy and urinary albumin excretion [44]. By contrast, excessively low levels of VEGF are associated with renal deterioration in the type 2 diabetic model [44,53], suggesting that a too-low VEGF level can be just as damaging as when the VEGF level is too high [53]. Interestingly, PPARα agonists have been shown to inhibit endothelial VEGFR-2 expression, and fenofibrate treatment induces a significant reduction of VEGF levels in serum [53,55]. Although the data regarding the effects of administration with PPARα agonists on VEGF changes in diabetic kidneys are lacking, angiogenic modulation and endothelial cell stabilization by PPARα agonists might be responsible for the potential renoprotective effects in diabetic nephropathy models. Recently, we found that dual VEGFR1 and VEGFR2 inhibition in db/db mice aggravated diabetic peripheral neuropathy, including a decrease in nerve conduction velocity and an increase in the tactile threshold of the sciatic nerve associated with vascular rarefaction resulting from endothelial cell apoptosis, which completely recovered to levels of non-diabetic db/m mice by fenofibrate (unpublished data).
As renal lipotoxicity can lead to chronic kidney disease, an overload of free fatty acid-bound albumin in the proximal epithelial cells induced tubular cell injury resulting from not only a decrease in the lipolytic enzymes but also increases in lipid accumulation and oxidative stress [56]. Fenofibrate, by contrast, inhibited palmitate-induced expressions of both monocyte chemoattractant protein-1 and PAI-1 and oxidative stress in the proximal tubular cells, which were associated with the overexpression of lipolytic enzymes and enhancement of renal lipolysis. Therefore, we need more experimental evidences using various types of renal cells including podocytes in order to investigate whether pharmacological activation of PPARα could be a therapeutically suitable strategy against glomerular and tubuleinterstitial lesions in diabetic nephropathy.
Clinical studies
Recent studies have shown a beneficial effect of fibrate treatment on type 2 diabetes and diabetic nephropathy [57-59]. In normotensive patients with non-insulin-dependent diabetes, effective treatment of dyslipidemia by a PPARα activator gemfibrozil for one year was associated with significant stabilization of urine albumin excretion [58]. The Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study reported that fenofibrate treatment was associated with reductions in cardiovascular disease outcomes [59]. This study included 9,795 patients with type 2 diabetes. The results demonstrated that fenofibrate treatment was associated with an 11% reduction in total cardiovascular disease events [59]. Interestingly, the FIELD study showed that fenofibrate resulted in significantly more patients' regressing or not progressing in their urinary albumin excretion. In the fenofibrate group, albuminuria progressed in 9.5% of the patients compared to 11.0% of patients in the placebo group. Albuminuria regressed in 9.4% of patients in the fenofibrate treatment group and in 8.2% in the placebo group participants [44,59]. The Action to Control Cardiovascular Risk in Diabetes (ACCORD) study also demonstrated that fibrate therapy with intensive glucose control could significantly reduce microalbuminuria (38.2% vs. 41.6%, P=0.01) and macroalbuminuria (10.5% vs. 12.3%, P=0.03) despite the marginal effects of fibrates in the management of dyslipidemia [60,61]. Unfortunately, most trials with PPARα agonists have been designed for cardiovascular disease, not renal disease, as a primary end point. The effect of fenofibrate on urinary protein excretion appears to be minor compared with the major effects on cardiovascular disease events [62]. Another weakness is that most studies have been limited to people with type 2 diabetes. In the future, more large-scale, prospective, randomized trials will be necessary to evaluate the efficacy of fibrates on renal outcomes in patients with type 1 or type 2 diabetes.
LIMITATIONS OF PPARα AGONISTS
Despite evidence of the beneficial effects of PPARα agonists in patients with diabetes, there are still many issues to be addressed concerning their safety in clinical use. The most important concern is due to the fact that fibrate treatment typically results in increased serum levels of creatinine and cystatin C and might potentially decrease the estimated glomerular filtration rate and creatinine clearance [44]. More care should be taken when prescribing fibrates to patients with mild-to-moderate renal insufficiency. Currently, it is recommended that fenofibrate dosages should be reduced by one third in chronic kidney disease (CKD) stage 2, by an additional one third in CKD stage 3 and 4, and avoided in CKD stage 5 [62]. Although the mechanisms of deterioration in renal function with fibrates are not clear, renal hemodynamic changes in association with the reduction of cylcooxygenase 2 levels, an inhibitory action of fenofibrate on the excretion of creatinine by the kidneys, or an increase in the flow of creatinine from muscle might be an explanation [44,63].
In addition, the fact that PPARα is subject to tissue-specific regulation should be considered. Over-expression of PPARα in the heart results in increased fatty acid oxidation, elevated lipid droplets, and worsened cardiomyopathy, suggesting that cardiac PPARα activation may be harmful [54,64]. By contrast, an increase in PPARα expression in the diabetic kidney is evident [49], indicating compensatory PPARα activation in response to the renal damage suffered. The proper balance between overactivation and underactivation of PPARα may differ in each type of affected organ or tissue. To maximize the therapeutic potential and minimize the harmful effects of PPARα, future investigations should examine the development of selective agents with tissue-, organ- or disease-specific effects and targeted gene-selective activities. This will require development of more selective PPARα modulators.
CONCLUSION
Recent studies have suggested beneficial roles for PPARα and PPARα target genes as therapeutic targets in the treatment of disorders involving inflammation, atherosclerosis, oxidative stress, angiogenesis, and RAS. PPARα agonists have traditionally been used to lower circulating fatty acids and TG. Currently, considerable evidence suggests that PPARα is involved in the pathogenesis of diabetic nephropathy and contributes to the extrametabolic control of renal function. Although the mechanisms of the beneficial effects of fibrates in the kidneys are still under investigation, PPARα would be a promising therapeutic target in the management of diabetes and diabetic nephropathy.
 XML Download
XML Download