

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
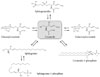
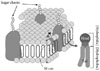
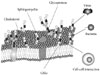
Glycosphingolipids (GSLs) are ubiquitous cell membrane components of eukaryotic cells [1,2]. They are amphipathic molecules consisting of a ceramide and sugar chains, including one or more glucose, galactose, and sialic acid residues, as shown Fig. 1. The ceramide portion is hydrophobic, and the sugar portion is hydrophilic as shown Fig. 2. These molecules are embedded in the leaflet of the membrane, as shown Fig. 3. GSLs are also present in intracellular components such as the Golgi apparatus, nuclear membrane, and mitochondria. GSLs, along with the associated membrane components, circulate through these organelles. In the Golgi apparatus, GSLs are newly synthesized by the addition of saccharides one by one. On the other hand, in lysosomes, GSLs are degraded by the removal of saccharides one by one. These products are the result of enzyme reactions [3]. More than 400 species of GSLs possessing different sugar structures have been reported, although only seven monosaccharides have mainly been found in vertebrate GSLs.
Microdomains called lipid rafts are functional units in cell membranes [4,5]. Such microdomains in cell membranes consisting of GSL-cholesterol, function as platforms for the attachment of lipid-modified proteins, such as glycosylphosphatidylinositol (GPI)-anchored proteins [6]. These specialized membrane microdomains may organize the assembly of signaling molecules, membrane protein trafficking, and regulate neurotransmission and receptor trafficking [6]. Rietveld and Simons [7] found lipid rafts in model membranes to be related to the immiscibility of ordered and disordered liquid phases. The cause of this immiscibility is uncertain, but the immiscibility may have a constant effect in events such as phosphorylation of the signal transduction molecules. These events include phosphorylation by tyrosine kinases and/or serine/threonine kinases. Lipid rafts have been found by researchers to be involved in many signal transduction processes, including T cell antigen receptor signaling [8], B cell antigen receptor signaling [9], epidermal growth factor receptor signaling [10], and insulin receptor signaling [11,12].
In this review, I summarize our understanding of the role of GSLs on the insulin receptor abnormality as one of the pathophysiology of diabetes, and rodent models for the research on insulin resistance.
CHARACTERIZATION OF GSLS
GSLs in a subclass that contains sialic acid residues are known as gangliosides, and are abundantly present in the nervous system. Other subclasses of GSLs such as the globo series and isoglobo series, are abundant in red blood cells and leukocytes, respectively. Neolacto series and asialo series are also known. The synthesis of many GSLs starts from ceramide. Glucosylceramide (GlcCer) synthase, the first enzyme required for the synthesis of GSLs, is a transmembrane protein located in the endoplasmic reticulum (ER) with its C-terminal catalytic domain located in the cytoplasm [13]. Following the synthesis of glucosylceramide, lactosylceramide, which consists of ceramide, glucose, and galactose, is synthesized in the Golgi apparatus as a key molecule in this synthetic pathway. Through this molecule, higher-order GSL structures are produced by a series of Golgi glycosyltransferases. As a regular pathway, GlcCer is synthesized on the cytosolic side and must translocate across to the Golgi lumen for LacCer synthesis. However, recent evidence has shown that GlcCer destined for glycolipid synthesis follows a different pathway and is transported back into the ER via the late Golgi protein FAPP2 [14]. GSLs have been implicated in many fundamental cellular processes, including cell growth, migration, differentiation, morphogenesis, early development, and functioning of the nervous system [15-20]. GSLs seem to construct plasma membrane microdomains, known as rafts and caveolae [21,22], which are rich in sphingolipids, cholesterol, and sphingomyelin. As for metabolism, Whitmore et al. [23] demonstrated significant cell-to-cell heterogeneity in the amounts of gangliosides and their metabolites. GSL expression varies among and within cell populations, during cellular differentiation, and between normal cells and their transformed counterparts. For the regulation of GSL metabolism, Memon et al. [24], have reported that endotoxins (lipopolysaccharide, LPS) and cytokines enhance hepatic sphingolipid synthesis, increase the activity and mRNA levels of serine palmitoyltransferase, the first committed step in sphingolipid synthesis, and increase the content of sphingomyelin, ceramide, and GlcCer in circulating lipoproteins of Syrian hamsters. Tettamanti described the metabolic turnover or recycling of GSLs in a review [25]. Briefly, the events to be considered are de novo biosynthesis, in situ chemical modifications at the plasma membrane level, direct recycling to the plasma membrane from early endosomes, sorting to the Golgi apparatus from endosomes with subsequent glycosylation, degradation at the lysosomal/late endosomal level, salvage pathways, and complete degradation to waste products. The half-lives of GSLs seems to range from 2 to 6.5 hours, to three days depending on the cells or tissues used.
GSLS, DIABETES, AND INSULIN RESISTANCE
Insulin is an anabolic hormone with regard to carbohydrate and lipid metabolism. Insulin exerts its effects via the insulin receptor (IR), a transmembrane receptor tyrosine kinase with two α-subunits and two β-subunits [26]. The α-subunits contain the insulin-binding sites. The β-subunits form transmembrane and intracellular parts of the receptor. Following insulin binding, the IR undergoes a conformational change that allows trans phosphorylation of the catalytic sites on multiple tyrosine residues [27]. The most important sites for the action of insulin are the liver, adipose tissues, and muscles. The concentration of IR is high in these tissues, with more than 200,000 receptors on adipocytes and hepatocytes [27]. On the other hand, insulin resistance is defined as the reduced ability of a cell to respond to physiological concentrations of insulin. As a result of insulin stimulation, insulin binds to the IR and induces autophosphorylation of tyrosine kinase under normal physiological conditions. Following autophosphorylation, at least two signaling cascades are known. In the first, Akt together with protein kinase C promotes translocation of the glucose transporter (GLUT-4) to the plasma membrane enabling the uptake of glucose in the bloodstream. In the second, the mitogen activated protein kinase cascade is initiated [28]. The PI(3)K/Akt signaling pathway is central to proper insulin signaling [29]. As for cholesterol, Simons and Toomre [30] had reported that cholesterol depletion inhibited raft-dependent signaling. Therefore, cholesterol in the cell membrane seems to act as a positive effector for IR and IRS-1 phosphorylation. What then are the effects of IR and IRS-1 phosphorylation by GSLs, which are components of the cell membrane like cholesterol? Generally, GSLs may play a prominent role in cell signaling, acting as both first and second messengers in a variety of signaling and regulatory pathways [31]. Of all the sphingolipids, ceramide and sphingosine, together with their phosphorylated counterparts, have received the most attention with regard to bioactivity. As one of the structures that GSLs participate in, lipid rafts are required for effective PI(3)K/Akt signaling. Moreover, Vainio et al. [32] have provided evidence that the IR is recruited to detergent-resistant domains upon ligand binding and the clustering of GM2 ganglioside inhibits IR signaling, apparently by excluding the ligand-bound IR from these domains. GM2 ganglioside may be important for the dynamic recruitment of the ligand-bound IR into the raft.
Diabetes mellitus type 2, which is known as non insulin-dependent diabetes mellitus or adult-onset diabetes, is a metabolic disorder that is characterized by a high glucose level in the bloodstream, insulin resistance, and insulin deficiency. Type 2 diabetes is a lifestyle- and genetic-related disease. The importance of various factors in the onset and pathological symptoms of the disease has been emphasized. There are many drugs with different mechanisms of action for this disease. For example, sulfonylureas lower blood sugar by stimulating the pancreas to release more insulin. Biguanides improve insulin's ability to move sugar into cells, especially into muscle cells. Thiazolidinediones improve insulin's effectiveness in muscle and fat tissues. Alpha-glucosidase inhibitors block enzymes that help digest starches, slowing the rise in blood sugar. Dipeptidyl peptidase IV inhibitors work to lower blood sugar in patients with type 2 diabetes by increasing insulin secretion from the pancreas and reducing sugar production. Studies have shown that some diabetes pills may help prevent diabetes and diabetes-related complications. On the hand, Ottenhoff et al. [33] reported that pharmacological inhibition of glucosylceramide synthase enhanced insulin sensitivity. They developed a highly specific, small molecule inhibitor of glucosylceramide synthase, N-(5'-adamantane-1'-yl-methoxy)-pentyl-1-deoxynojirimycin (AMP-DNM). AMP-DNM counteracts TNF induced abnormalities in GSL concentrations and concomitantly reverses abnormalities in insulin signal transduction. Due to the pharmacological actions mentioned above, AMP-DNM significantly reduces GSL but not ceramide, concentrations in various tissues in mice and rats. Treatment of ob/ob mice with AMP-DNM normalizes their elevated tissue glucosylceramide levels, markedly lowers circulating glucose levels, improves oral glucose tolerance, reduces glycated hemoglobin, and improves insulin sensitivity in muscle and liver tissues. It is typically reported that the density of GSLs is closely related to insulin sensitivity in vivo. D-threo-1-Phenyl-2-decanoylamino-3-morpholino-1-propanol (D-PDMP) is a well-known inhibitor of UDP-glucose, which is the first step of GSL synthesis [34]. This inhibitor has long been employed to study the roles of GSLs. The inhibitor is transported to the ER and Golgi apparatus [35]. Following treatment with D-PDMP, nearly all GSL production is dramatically reduced on the surfaces of cells. In some cell lines, exogenously added Lac-Cer rescues blocking by D-PDMP.
ANIMAL MODELS OF INSULIN RESISTANCE
Rat models
Though many animal models of diabetes have been established, it is impossible to completely reproduce the condition of human patients. However, the knowledge gained from model animals contributes to our understanding of genetic diseases, the effects of drugs, elucidation of pathological conditions, prevention, and treatments. Diabetes is a result of the lack of insulin. Amongst rodents, high-calorie feeding can induce hypertriglyceridemia, insulin resistance, and hypertension. Sprague-Dawley (SD) rats [36] and Wistar rats [37,38] are established models of sucrose-induced insulin resistance and hypertriglyceridemia. There are also high-fat rat models. The Goto-Gakizaki (GK) rat was established by genetic segregation based on the glucose level as early as eight days after birth. In GK rats, blood glucose level after oral glucose administration is high, with a severe glucose tolerance abnormality. GK rats also have metabolic-endocrine abnormalities, such as hyperglycemia and insulin resistance. In the pancreas, islet deformation, secretion abnormality, and gradual b-cell loss were observed [39]. The diabetic Zuker fatty rat was established as a fatty model rat [40,41]. An abnormal gain of weight is observed three weeks after birth and the body weight increases with age. Obese Zucker rats have high levels of lipids in their blood and are resistant to insulin. The Otsuka Long Evans Tokushima fatty (OLETF) rat is a useful animal model of type II diabetes with obesity [42,43]. The OLETF rat presents clinically relevant phenotypes of diabetes such as hyperinsulinemia, hyperglycemia, insulin resistance, hypertriglycemia, and mild obesity. In vivo insulin-stimulated glucose uptake as measured with a euglycemic clamp was reduced 37% compared with that in non-diabetic control rats. Morphological studies on the OLETF rat pancreas showed enlarged multi-lobulated fibrotic islets, and hyper states of insulin production and secretion. The Wister fatty rat (fa/fa) was developed by taking the fatty gene from the Zucker rat and transferring it to the Wistar-Kyoto rat, which is less sensitive to insulin, and less tolerant to glucose, than lean Zucker rats. Wistar fatty rats also exhibit obesity, hyperphagia, hyperlipidemia, hyperinsulinemia, and peripheral insulin resistance, similar to Zucker rats. The tolerance and insulin response to oral glucose are decreased with advancing age in males. Hypertrophy of pancreatic islets and degranulation of beta cells have also been observed.
Mouse models
In contrast to rats, mice are used less frequently as a model for sucrose/fructose-induced insulin resistance and hypertriglyceridemia. The response to high fructose/sucrose diets is very strain-dependent in mice [44], and commonly used strains like C57Bl/6 either do not develop insulin resistance or only develop it slowly [45]. When C57Bl/6 mice were fed a high-fructose diet for eight weeks, they developed increased mean arterial pressure, reduced glucose tolerance, and increased plasma cholesterol that was attributed to the activation of the sympathetic and angiotensin systems [46]. Like the high dietary fructose used for the induction of diabetes, fat-based diets can induce diabetes and obesity [47]. The effects of fat and sucrose were compared separately and in combination in diabetes- and obesity-prone C57Bl/6J and diabetes- and obesity-resistant A/J mice. After four months, the feed efficiency (weight gained divided by calories consumed), did not differ across diets in A/J mice. However, C57Bl/6J mice had significantly increased feed efficiency for the fat-based diet. The KK mouse was established in 1962 [48]. It shows a striking intolerance to glucose, hyperinsulinism, and obesity. Of particular interest is the development of mild to moderate glomerulosclerosis in the prediabetic stage. Later in life, progression to severe glomerulosclerosis and attendant proteinuria are observed [49]. The ob/ob mouse is a mutant strain that eats excessively and becomes obese. It is one of the typical animal models of type 2 diabetes. In adipose tissue, enlargement of cells and an increase of cell number were observed. Idenfication of a mutation in the ob gene led to the discovery of leptin, which is important for the control of food consumption. Since the ob/ob mouse cannot produce leptin, its food intake is uncontrolled [50]. A mouse similar to the ob/ob mouse, the db/db mouse, was eatablished [51]. This mouse exhibits severe diabetes with longstanding hyperglycemia. Moreover, within six weeks of age, db/db mice develop significant obesity, fasting hyperglycemia, and hyperinsulinemia. The db/db mice have a prominent peak in the low density lipoprotein (LDL) range. Upon consumption of a 0.15% (wt/wt) cholesterol and 21% (wt/wt) fat "western" diet, db/db mice show elevated plasma cholesterol [52]. The non-obese diabetic (NOD) mouse was established in 1984. Diabetes develops in NOD mice as a result of insulitis, leukocytic infiltration of the pancreatic islets [53,54]. The incidences of spontaneous diabetes in the NOD mouse are 60% to 80% in females and 20% to 30% in males. The onset of diabetes also varies between males and females. The time difference of the onset may be attributed to the breeding environment. For chemical induction of diabetes, the N-nitro derivative of glucosamin (STZ) is used frequently. STZ is a glucosamine-nitrosourea compound that is particularly toxic to the insulin-producing β-cells in the pancreas in mammals. Multiple small doses of STZ produce a delayed, progressive increase in plasma glucose in mice within 5 to 6 days after the injections. High dose injection of STZ results in acute hyperglycemia. Alloxan, a crystalline substance produced by oxidation of uric acid and a toxic glucose analogue, selectively destroys β-cells in the pancreas when administrated to laboratory animals, including mice and rats [55]. This causes insulin-dependent diabetes mellitus similar to type 1 diabetes in human patients. This most likely occurs because of selective uptake of the compound due to its structual similarity to glucose, as well as the β-cell's highly efficient uptake mechanism [56].
Gene-manipulated mouse models
Knockout mouse technology is a valuable research tool and contributes to the clarification of biological questions. Via disruption of a specific gene in the mouse and observation of differences from normal behaviour or responses, researchers can infer its probable biological function. However, some limitations exist. With knockout mice, it is often difficult to determine a gene's function in relation to human diseases as the genes in mice may have different functions from the human genes. Investigators have established knockout mouse models for diabetes research. However mice carrying a targeted disruption of the diabetes-related gene might develop early onset diabetes and die perinatally. Detailed examination of the pancreas revealed proliferation arrest and cell death in pancreatic β-cells. As a consequence, it is likely that these cumulative effects cause diabetes.
Considering carbohydrate chains including GSLs, diabetic mice with P-selectin glycoprotein ligand-1 (PSGL-1) deficiency show a protective phenotype against obesity-related insulin resistance [57]. PSGL-1 knockout mice fed a high-fat diet (HFD) show a remarkable reduction of macrophage accumulation and expression of pro-inflammatory genes. Moreover, adipocyte hypertrophy, insulin resistance, lipid metabolism, and hepatic fatty change were improved in PSGL-1 knockout mice compared with WT mice fed HFD. PSGL-1 is a crucial adhesion molecule for the recruitment of monocytes into adipose tissues in obese mice. Several studies have reported that blockade of PSGL-1 reduces inflammatory reactions [58,59]. Mice with gene disruption of GM3 are viable without major abnormalities, but have heightened sensitivity to insulin [60]. The basis for the increased insulin sensitivity in mutant mice was found to be enhanced IR phosphorylation in skeletal muscle. Importantly, the absence of the ganglioside GM3 confers protection from HDF-induced insulin resistance to the knockout mice. Gganglioside GM3 is a negative regulator of insulin signaling.
CONCLUSION
In this review, I summarized the molecular-based diversity of GSLs. Microdomains, termed lipid rafts, which are special structures of cell membranes are made of GSLs and protein receptors. These specialized membrane microdomains may compartmentalize cellular processes and function for the assembly of signaling molecules. Since many molecules, such as receptors and channels penetrate cell membranes, the positive or negative influence (e.g., regulation of signaling, internalization, and trafficking) of the cell membrane, including GSLs and other components, such as cholesterol, and sphingomyelin, is not small. On the other hand, model animals including mice with gene disruption are used for diabetes research. They provide an essential research tool for academic institutions and commercial companies by enabling the rapid screening of novel compounds, and testing of new gene-based therapies for the treatment of diabetes and diabetes-related symptoms.
 XML Download
XML Download