

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Diazepam is a benzodiazepine agent potentiating the inhibitory effect of γ-aminobutyric acid (GABA) by enhancing the membrane permeability of chloride ions. It is commonly used as adjunctive therapy for various psychiatric disorders such as anxiety, alcohol withdrawal, skeletal muscle cramps, and seizures. The unbound fraction of diazepam in plasma is approximately 2% with the blood-to-plasma concentration ratio of approximately 0.58.[1] Diazepam is metabolized to temazepam and nordiazepam, which are ultimately converted to oxazepam by CYP3A and CYP2C19 isoforms. Oxazepam and temazepam are further metabolized to glucuronide conjugates.[2]
Previous studies have proposed several analytical methods using high-performance liquid chromatography (HPLC) with an ultraviolet (UV) [34] or mass spectrometry (MS) [56789] detection to determine the concentrations of diazepam administered by various routes including nasal spray, rectal gel, intramuscular injection, and oral formulations.[101112131415] However, these previously developed analytical methods have been associated with several limitations including inadequate sample preparation due to difficulty in removing impurities,[5] inadequate sensitivity (a lower limit of quantitation [LLOQ] up to 50 ng/mL),[6] labor-intensive sample preparation process,[7] the need for a relatively large volume of plasma (≥ 100 µL) for analysis,[8] and long retention times (≥ 8 min).[9] Therefore, a new bioanalytical method to quantitate diazepam has been needed to improve those limitations.
The objective of this study was to develop and validate a rapid, selective, and sensitive HPLC-MS/MS method for the analysis of diazepam in human plasma. The applicability of the diazepam bioanalytical method developed in this study was evaluated in a bioequivalence study where more than 1300 human plasma samples were collected from 40 healthy Korean male volunteers receiving two different formulations of a diazepam 2-mg tablet as a single dose.
Methods
Chemicals and Reagents
Chemicals and Reagents Diazepam (99.8% purity) was supplied by the Ministry of Food and Drug Safety (MFDS) (Cheongju, Korea). Voriconazole (99.6% purity) was purchased from Sigma Aldrich (Saint Louis, MO, USA). Figure 1 shows the molecular structures of diazepam and voriconazole, respectively. Ethyl acetate, methanol, and n-hexane were obtained from J.T. Baker (Philipsburg, NJ, USA). Water was purified by using a Milli-Q® water purification system (Millipore Co., Milford, MA, USA).
Hplc-ms/ms conditions of diazepam analysis
Chromatographic separation was performed using Shimadzu Nexera X2 (Shimadzu, Japan) equipped with an autosampler, a column heater, and a binary pump. Utilizing the Cadenza CD-C18 column (150 × 3.0 mm, 3 µm) (Imtakt, Japan) at 40℃, the analytes were separated using the isocratic mobile phase of 10 mM ammonium acetate:100% methanol (5:95, v/v) at a flow rate of 0.4 mL/min. The column was eluted into the Applied Biosystems MDS SCIEX API 4000 triple quadrupole mass spectrometer (Applied Biosystems, Canada) with an ESI source, operating in the multiple reaction monitoring (MRM) mode under unit mass resolution conditions in mass analyzers. The optimized MS condition was as follows: curtain gas flow 20 L/h, collision gas flow 6 L/h, collision energy 45 V, and declustering potential 51 V. Data analysis was performed using Analyst 1.6 software (Applied Biosystems, Canada).
Preparation of Calibration Standard and Quality Control-Samples of Diazepam
Standard stock solutions (1 mg/mL) of diazepam and voriconazole (internal standard [IS]) were prepared in 100% methanol and stored in the refrigerator at −20℃. Working solutions of diazepam for calibrations were obtained by serial dilution with 50% methanol from the stock solution to concentrations of 5, 10, 50, 100, 500, 1,000, and 3,000 ng/mL. Calibration standards and QC samples of diazepam in plasma were prepared by diluting the corresponding working solutions with blank human plasma. The final concentrations of calibration standards were 0.5, 1, 5, 10, 50, 100, and 300 ng/mL. QC samples were prepared at three different concentrations of 1.5, 100, and 240 ng/mL. The IS working solution was prepared at 200 ng/mL. All solutions were stored in the −20℃ freezer when not in use.
Sample Preparation
The human plasma samples stored in −70℃ were thawed at room temperature. An aliquot of each sample (50 µL) was placed into a polypropylene microtube, and IS solution (20 µL, 200 ng/mL), 0.1% formic acid (100 µL), and 1.2 mL of ethyl acetate: n-hexane (80:20, v/v) were added. The mixture was vortexed for 10 min and then centrifuged at 14,000 rpm for 10 min. The supernatant (1 mL) was transferred to a new microtube and evaporated under nitrogen gas at 40℃. The remaining residue was reconstituted with 300 µL of 90% methanol. For the analysis, 6 µL supernatant aliquot was pipetted into a vial and then injected into the HPLC-MS/MS system.
Validation of the Analytical Method
The developed analytical method was validated for selectivity, linearity, accuracy, precision, recovery, matrix effect, and stability according to the bioanalytical method validation guidelines by the MFDS and the USFDA.[1617]
Specificity and Lower Limit of Quantitation
To evaluate its specificity, the analytical method was assessed by comparing the chromatograms obtained from the samples containing diazepam and IS with blank samples. Six blank human plasma samples from different volunteers were collected under equivalent controlled conditions, and they were subjected to the sample preparation procedure as described above. Afterward, they were evaluated for the interference of the endogenous plasma components with diazepam or IS. The lower limit of the quantification (LLOQ) was defined as the lowest plasma diazepam concentration corresponding to a signal-to-noise (S/N) ratio of ≥ 10 with an acceptable precision not exceeding 20% and accuracy within ± 20%.
Linearity
Linearity was evaluated using a weighting linear least-squares regression (1/χ2) model over the diazepam concentration range of 0.5–300 ng/mL. The calibration curves were y = ax + b, where y is the mean of the peak area ratios of the analytes to their internal standards, a is the slope of the calibration curve, b is the y-axis intercept of the calibration curve, and x is the analyte concentration. A coefficient of determination (r2) of 0.99 or greater was considered satisfactory.
Precision and Accuracy
In order to assess inter- and intra-day precision and accuracy of the analytical method, four concentrations of QC samples were analyzed. Intra-day assays were performed on five replicates per day, and inter-day assays were carried out for three consecutive days. The acceptance criteria for the accuracy and precision based on each back-calculated standard concentration were a 15% coefficient of variation (CV; standard deviation/mean × 100) from the nominal value except at the LLOQ, where the acceptance limit of %CV was 20%.
Stability
The stock solution stability of diazepam was tested using three replicates of low and high QC at room temperature for 3 h and −20℃ for 93 days. The stability of the analyte in human plasma was evaluated using low, middle, and high QC samples under the following conditions: refrigerated (4℃), frozen (−70℃), room temperature (7 h), five freezing-thawing cycles (−70℃ to 20℃), and long-term frozen storage (−70℃) for 86 days.
Recovery and Matrix effect
Diazepam recovery in plasma was evaluated at three QC concentrations by comparing the analytical peak area of the pre-extracted QC sample and that of the post-extraction QC samples, which were prepared by adding compound to post-extracted drug-free plasma at each concentration. The matrix effect was assessed by comparing the peak areas at the QC concentrations spiked after extraction into plasma with standard solutions of the analyte.
Application to a Bioequivalence Study
Our validated analytical method was applied to a cross-over bioequivalence study to determine the concentrations of diazepam in human plasma samples collected from 40 healthy Korean male volunteers who received a single-dose 2 mg diazepam tablet (Daewon [DW] or Samjin [SJ]) orally. This study was approved by the MFDS as well as the institutional review board (IRB) at the study site, and written informed consent was obtained from study participants in accordance with the principles of the Helsinki Declaration (World Medical Association Declaration of Helsinki 2000) [18]. Subjects were instructed not to drink or smoke until 24 h before the study. Blood samples (7 mL) were collected in heparinized tubes as scheduled at the following times: prior tz and harvested plasma was stored at −70 ℃ until analysis. Cmax (peak plasma concentration) and Tmax (time to reach Cmax) of diazepam were determined based on the individual plasma concentration-time profiles. A non-compartmental method for extravascular input, provided in BA Calc 2007 software, was used to calculate pharmacokinetic parameters including AUClast (area under the plasma drug concentration-time curve between 0 and the last measurable time point) and extrapolated AUCinf (AUC from 0 to infinity). Bioequivalence of the two diazepam tablets was evaluated based on the ratio (test/reference) of log-transformed data (Cmax and AUClast) with 90% confidence intervals (CIs) using the K-BE test II software distributed by the MFDS.[17]
Results
Method development
Mass spectrometry
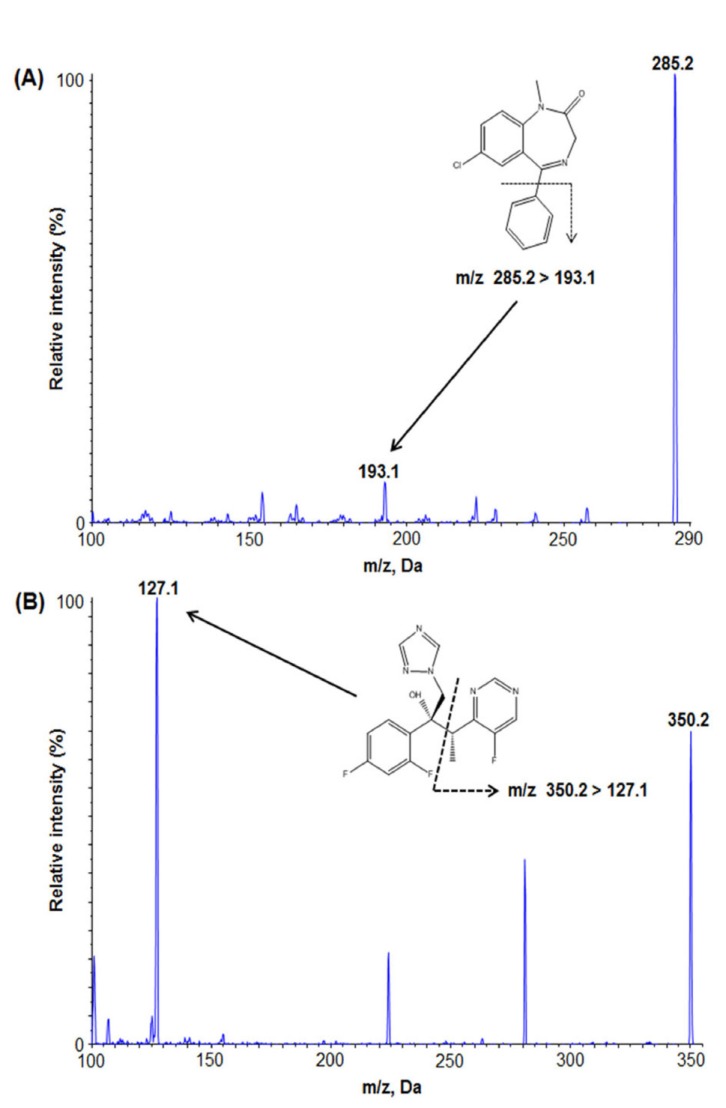
Diazepam and voriconazole (IS) were dissolved in 100% methanol at a concentration of 250 ng/mL. The samples were injected into a mass spectrometer with a syringe pump operating at a flow rate of 10 µL/min. The maximum amount of product ion was obtained in a positive mode using a turboionspray ESI interface. Based on the Q1 full scan, diazepam and IS were detected at m/z transition of 285.2→193.1 and 350.2→127.1, respectively (Fig. 1).
Chromatographic conditions
Several HPLC methods were investigated using different columns and mobile phases to reduce ion suppression induced by endogenous substances and to optimize the peak shapes and retention times of diazepam and IS. In our preliminary study, Phenomenex Luna HILIC (50 × 2.0 mm, 3 µm), Cadenza CD-C18 (150 × 3.0 mm, 3 µm), Phenomenex Luna C8 (50 × 2.0 mm, 3 µm), and Halo™ phenyl hexyl (2.1 × 150 mm, 2.7 µm) columns were tested for optimization. The best results in terms of separation, peak shape, and reproducibility were obtained with a Cadenza CD-C18 column (150 × 3.0 mm, 3 µm); when using other columns, peak tailing or fronting occurred. For the mobile phase, a series of aqueous mobile phases containing different additives (acidic buffers and additives such as formic acid, ammonium acetate, and ammonium formate) with different pH values were tested with methanol solution. A mobile phase consisting of 10 mM ammonium acetate (pH 3.0, adjusted with formic acid) in methanol (5:95, v/v) achieved symmetric peak shapes and short retention time (3 min) (Fig. 2).
Method Validation
Specificity and Lower Limit of Quantitation
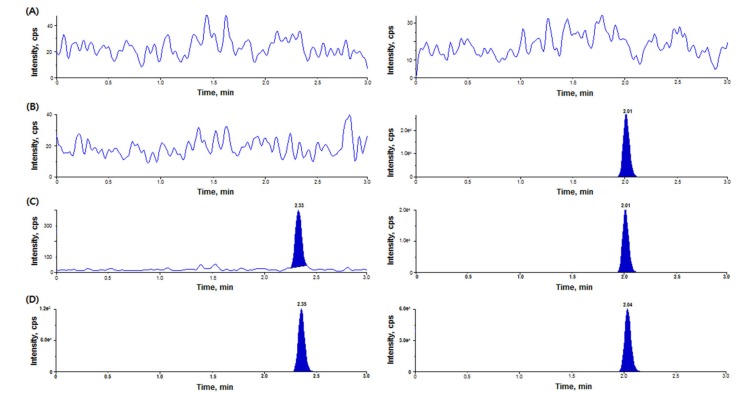
Under the experimental conditions described above, Figure 2 showed the chromatograms of blank plasma, plasma sample spiked with IS only, plasma sample spiked with diazepam at the LLOQ (0.5 ng/mL), and plasma sample spiked with IS from a volunteer at 0.75 h after an oral administration of 2 mg diazepam. No interference was observed with diazepam and IS. The S/N ratio at the LLOQ (0.5 ng/mL) was > 10. The LLOQ for our method was lower than that for previously developed methods using LC-MS/MS.[569] The precision and accuracy of several LLOQ samples were adequate and applicable to bioavailability studies.
Linearity
The regression equation with mean ± standard deviation regression coefficient for the calibration curve (n = 5) was y = 0.0246 (± 0.002)x − 0.000559 for diazepam with the correlation coefficients > 0.99 for all curves, suggesting adequate linearity and reproducibility over the concentration range.
Precision and Accuracy
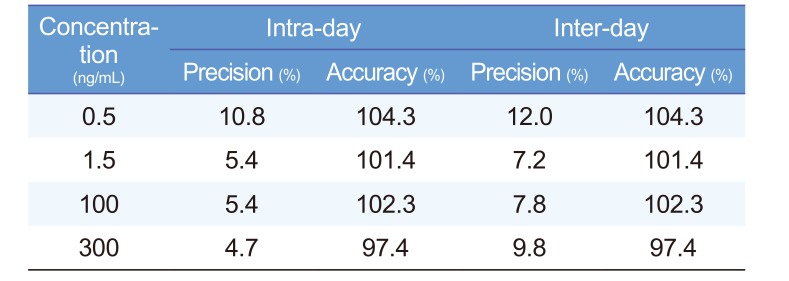
Intra-day and inter-day precision and accuracy for diazepam in human plasma at four concentrations are summarized in Table 1. Intra-day precision and accuracy ranged from 1.79% to 12.47% and from 86.87% to 112.92%, respectively. Inter-day precision and accuracy ranged from 7.25% to 11.98% and from 97.37% to 104.25%, respectively. Thus, our precision and accuracy results were within the acceptable criteria based on the MFDS guideline, suggesting adequate precision and accuracy of our developed analytical method.[17]
Stability
Table 2 presents the stability of diazepam in stock solutions using two QC concentrations (1.5 and 240 ng/mL) and in human plasma using three QC concentrations (1.5, 100, and 240 ng/mL) under different conditions during sample preparation and storage. Diazepam was considered stable in stock solutions and human plasma with the concentration deviation from the newly prepared sample < ± 15%. Our diazepam stability test results demonstrated a good stability without any significant degradation of diazepam over all steps of the routine sample preparation and storage for pharmacokinetic and bioavailability studies.
Recovery and Matrix effect
To remove interfering substances and decrease the LLOQ, the sample preparation procedures must be highly reproducible with high recovery of analyte and a minimum number of working steps. In this study, five different extraction solvents including methyl tert-butyl ether (MTBE), ethyl acetate, methyl chloride, n-hexane, and a mixture of ethyl acetate and n-hexane (80:20, v/v) were evaluated. The mixture of ethyl acetate and n-hexane (80:20, v/v) was chosen based on the adequate sensitivity and recovery of analytes. Although protein precipitation (PP) and solid phase extraction (SPE) methods have been previously used to extract the analyte, these methods are laborious and thus, may not be the best for routine analysis when processing a large number of plasma samples.[678] The extraction recoveries and matrix effects of diazepam following extraction at three QC concentrations were 81.92–86.63% and 88.15–93.54% (n = 6), respectively (Table 3). The extraction recovery and absolute matrix effect of IS were 87.61% and 89.71%, respectively, at 200 ng/mL (Table 3). These results suggested no endogenous substances substantially associated with the ion suppression in this analytical method, demonstrating the reliability of our bioanalytical method.
Clinical application
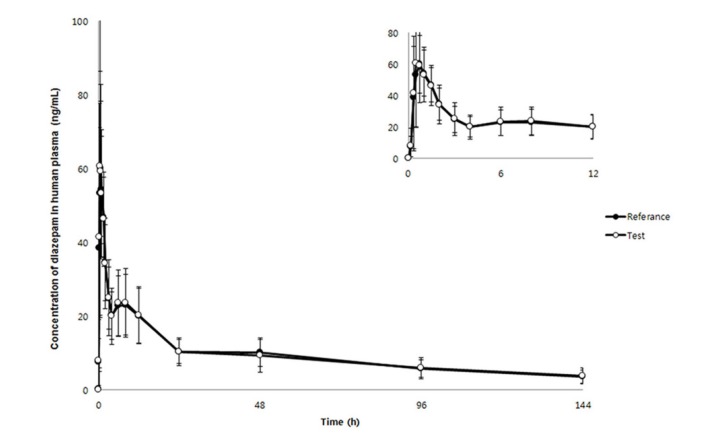
Our proposed HPLC-MS/MS method was successfully applied to a bioequivalence study of a single-dose, oral 2 mg diazepam tablet in 40 healthy male Korean volunteers to quantitate diazepam concentrations (n = 1300). The mean plasma diazepam concentration-time curve after an oral administration of a single-dose reference and test diazepam tablet (2 mg) is shown in Figure 3. No significant sequence effect was observed for any pharmacokinetic parameters. Compared with previous diazepam pharmacokinetic studies after an oral administration (AUCinf: 2760 ± 800 ng·h/mL [Dose: 5 mg] and 7551 ± 642 ng·h/mL [Dose: 10 mg]; Cmax: 131 ± 37 ng/mL [Dose: 5 mg] and 394 ± 19 ng/mL [Dose: 10 mg]), our study reported similar estimates of pharmacokinetic parameters (Table 4).[1920] The mean ± standard deviation (SD) AUCinf, AUClast, and Cmax were 1793.854 ± 969.919 ng·h/mL, 1347.273 ± 470.211 ng·h/mL, and 75.160 ± 21.68 ng/mL, respectively, for the reference drug (Table 4). For the test drug, the mean ± SD AUCinf, AUClast, and Cmax were 1796.746 ± 888.349 ng·h/L, 1343.681 ± 500.314 ng·h/mL, and 77.924 ± 30.76 ng/mL, respectively (Table 4). The 90% CIs for the geometric mean ratios of AUClast and Cmax were 0.96–1.03 and 0.93–1.10, respectively, satisfying the bioequivalence criteria based on the 90% CI for the geometric mean ratios of AUC and Cmax of 0.80-1.25.[1617] Therefore, the two diazepam tablets evaluated in this study were bioequivalent, no significant difference in the systemic disposition of diazepam between the two formulations.
Discussion
In this study, we described a reliable and efficient method to determine diazepam concentrations in human plasma. The full validation procedures demonstrated our proposed method, with a total running time of 3 min for each sample, was specific, rapid, and reproducible over the concentration range of 0.5–300 ng/mL. In previous studies to develop a bioanalytical method to quantitate diazepam in human plasma, samples were prepared using the protein precipitation (PP) method or solid-phase extraction (SPE) method.[5678] However, due to insufficient removal of impurities from the sample when using the PP method, the analytical method with samples prepared using the PP method may not be appropriate for the routine analysis of a large number of samples.[56] The SPE method is a complicating and time-consuming sample preparation procedure.[78] Therefore, in this study, a liquid-liquid extraction (LLE) method was used to effectively remove endogenous substances and thus, improve the efficiency of analyzing a large number of samples for human pharmacokinetic research.
Our developed method satisfied all of the MFDS and USFDA guidelines for validating a bioanalytical assay.[1617] Additionally, this method has been successfully applied to a bioequivalence study of diazepam to analyze more than 1300 clinical samples from healthy volunteers. Therefore, the diazepam bioanalytical method developed in this study may be more appropriate for the routine quantification of a large number of samples with an improved analytical sensitivity.
 XML Download
XML Download