

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
The therapy of central nervous system disorders is complicated by the fact that many drugs either do not cross the blood-brain barrier or, like penicillin, are actively transported from the cerebrospinal fluid (CSF) by the choroid plexus. As a result, a wide variety of antibiotic.[1] antineoplastic.[2] analgesic.[3] and antispasticity[4] drugs have been administered directly into the CSF. With the exception of morphine, which has been administered by the ICV route to patients with severe pain.[5] most analgesic and antispasticity drugs are administered intrathecally into the CSF surrounding the spinal cord. On the other hand, the ICV route is preferred for antibiotics and antineoplastic drugs because administration via this route ensures a more homogeneous drug distribution throughout the CSF space.[2] Rational ICV therapy requires an understanding of basic CSF physiology, the pathophysiology of the condition being treated, and the properties of the drug being used. In some cases, pharmacokinetic studies provide a means of combining these considerations so as to optimize therapy.
Background
The existence of CSF was known to both Hippocrates and Galen.[6] However, in subsequent autopsy technique the head was severed from the neck. This resulted in the loss of both blood and CSF from the brain and obscured the recognition of CSF for many centuries. Swedenborg[6] is credited with the discovery of CSF which he encountered during his search for the central nervous system (CNS) seat of the soul. However, it was only in the twentieth century that investigations led to our current understanding of CSF physiology and pathology. The early findings regarding CSF production, circulation, and clearance were summarized by Harvey Cushing[7] in his Cameron Lectures and termed the “third circulation”. Cushing identified the choroid plexus and arachnoid villi as the respective sites of CSF production and clearance and laid the groundwork for contemporary investigations.
Underlying Physiology
The CSF volume has been found to average 150 mL in adults. [8] As much as 80% of CSF is estimated to be produced by the choroid plexus of the lateral, third, and fourth ventricles. Total CSF production ranges from 400 to 600 mL/day and as much as 80% is produced by the choroid plexus in the lateral, third, and fourth ventricles. CSF is returned to the venous system via cranial arachnoid granulations that function as finger valves, permitting CSF drainage when CSF pressure exceeds that of the internal jugular system. Fluid circulation within the CSF space is largely dependent on the transmitted force of arterial pulsations but is also influenced by posture, respiration, and jugular venous pressure.[8]
The brain interstitial space is a second compartment that communicates with the CSF. It occupies roughly 15%-20% of total brain volume and is composed of interstitial fluid (ISF) and extracellular matrix.[9] Because the interstitial space is very narrow and tortuous, diffusion within the ISF is “hindered” with apparent diffusion coefficients being 30% to 60% lower than their expected free-water values. An estimated 20% of total CSF production is generated in the interstitial space by extrachoroidal secretion and cerebral capillaries that cross the blood-brain barrier. This results in bulk flow of newly generated CSF that is free of contamination by neural tissue excretory products. [10] ISF drainage occurs via the CSF and also by convective flow along what has been termed the glymphatic system. This system consists of a periarterial pathway that leads through the brain parenchyma to perivenous drainage pathways that are connected to the extracranial lymphatic system.[11] Astrocytic water channels, containing aquaporin-4 (AQP4), provide a low-resistance pathway for fluid movement along this system and help maintain convective flow between the paravascular spaces and the ISF.[12]
The Choroid Plexus
The choroid plexus consists of a monolayer of cuboidal epithelial cells that surround a core of connective tissue and capillaries.[13] The capillaries are fenestrated and permeable but the cells have apical tight junctions that constitute a blood-CSF barrier. The process of CSF formation is mainly due to the active secretion of sodium ions by sodium-potassium adenosine triphosphatase (Na+/K+-ATPase) and bicarbonate production by carbonic anhydrase. The action of these enzymes creates an osmotic gradient that drives water into the CSF.
The choroid plexus epithelial cells also express a wide range of other ion channels and pumps that contribute to CSF formation, as well as a number of transporters located in both the apical and basolateral cell regions that have pharmacologic significance.[13] Among the efflux transporters, the apical region contains high levels of organic acid transporter 3 (OAT3), which transports penicillin from the CSF, and peptide transporter 2 (PEP2). P-Glycoprotein is also expressed at the apical surface but it transports substances back into the CSF. The multidrug resistance proteins (MRPs), MRP1 and MRP4 and breast cancer resistance protein (BCRP) are expressed at the basolateral epithelial cell membrane and may reduce the availability of antiretroviral drugs within the CSF. A facilitated diffusion transporter for glucose (GLUT-1) is located in the basolateral region of choroid plexus epithelial cells. But in contrast to brain endothelium, it is not present in the capillaries that perfuse the choroid plexus.[14]
The choroid plexus also contains the highest levels of drug metabolizing enzymes in the CNS.[15] Human data is not available, but in rats the activity of epoxide hydrolase and uridine diphosphate glucuronosyltransferase (UGT) in the choroid plexus equals or exceeds that of the liver.[15] A number of cytochrome P-450 (CYP-450) enzymes are also present in choroid epithelial cells but their activity is less than in the liver. In addition, choroid epithelial cells contain all three cytosolic glutathione sulfotransferases and monamine oxidase.[15] These enzymes act as an effective metabolic blood-CSF barrier with the efflux of their products being transferred to the systemic circulation by transporters at the basal epithelial membrane.
The blood-CSF barrier is completed by a layer of arachnoid barrier cells that have tight junctions and rest on a continuous basal lamina that faces the CSF.[16] These cells express P-glycoprotein in their apical region and are unique in expressing BCRP at both their basal and apical membranes. They also contain many of the transporters and CYP‐450 enzymes that are present in choroid plexus epithelial cells. GLUT-1 and related GLUT transporters were not looked for in this study but are abundant in brain microvascular endothelium and in the ependymal cells that line the cerebral ventricular system.[17] These transporters maintain the ratio of CSF/blood glucose within a normal range of 45% to 65% under quasi-steady-state conditions (e.g. fasting)[18] but transport has been shown in dogs to be saturable at supraphysiologic blood glucose concentrations. [19] Glucose transfer between blood and CSF is bidirectional and the concentration difference results from CSF bulk flow. The CSF/blood glucose ratio is depressed in patients with cryptococcal meningitis due to impaired transporter function and its rate of return to normal has been used as a biomarker to monitor patient response to therapy.[20]
Pharmacokinetics of ICV Drug Administration
Despite the wide variety of therapeutic agents that have been administered by the ICV route, there are very few rigorous studies that describe their CSF pharmacokinetics. Despite the paucity of these studies, CSF drug concentrations have been used in many cases to guide individual patient dosing. For antineoplastic drugs, Bleyer et al.[21] have introduced the “concentration x time” (CXT) strategy that recommends maintaining CSF drug levels above the anticipated in vitro tumoricidal concentration. Similarly, the Infectious Disease Society of America guidelines for ICV therapy of patients with meningitis recommend that trough antibiotic levels be 10 to 20 times higher than the minimum inhibitory concentration of the isolated pathogenic organism.[22] Because aminoglycosides have concentration-dependent bactericidal activity and demonstrate a prolonged post-antibiotic effect, it is reasonable to design dose regimens that are based on these characteristics. Thus, Brown et al.[23] recommend adjusting ICV gentamicin doses to provide CSF peak levels between 15 and 20 µg/mL and trough concentrations ≤ 2 µg/mL.
Even when sound pharmacokinetic studies are available for guidance, the expected pharmacokinetic parameters may be altered in patients whose underlying disease has blocked normal CSF distribution and flow. This complication is particularly likely to occur in patients with neoplastic meningeal disease. Mason et al.[24] simultaneously injected 111indium-diethylenetriamine pentaacetic acid (DPTA) and methotrexate by the ICV route and used a scintillation gamma camera to monitor CSF flow in 17 patients with meningeal metastasis. The distribution of both compounds was restricted or delayed in 13 patients and blockage could usually be correlated with abnormalities that were identified by magnetic resonance imaging. Methotrexate therapy in these patients was generally ineffective and more likely to result in toxicity, so the recommendation was made that radionucleotide flow studies be used to screen patients with meningeal metastatic disease before beginning ICV chemotherapy. Impeded CSF flow is less likely to occur in patients with infectious meningitis. So it is possible to design initial therapeutic regimens for these patients on sound pharmacokinetic principles, with subsequent dose adjustments being based on measured CSF drug concentrations and the antibiotic sensitivity of the infecting organism.
Amphotericin B - An Historical Perspective
Before the introduction of amphotericin B in 1958, cryptococcal meningitis was uniformly fatal.[25] With the availability of this antifungal drug, the National Institute of Allergy and Infectious Diseases at the National Institutes of Health (NIH) launched a nationwide program in which patients with this disease were brought to the NIH Clinical Center for treatment. Amphotericin was administered intravenously to all patients. In addition, the most severely ill patients were treated intrathecally with amphotericin B injections into the lumbar subarachnoid space, with doses ranging from 0.25 to 1.0 mg.[26] These injections commonly resulted in adverse reactions that included back and leg pain, difficulty voiding, and temporary paresis.
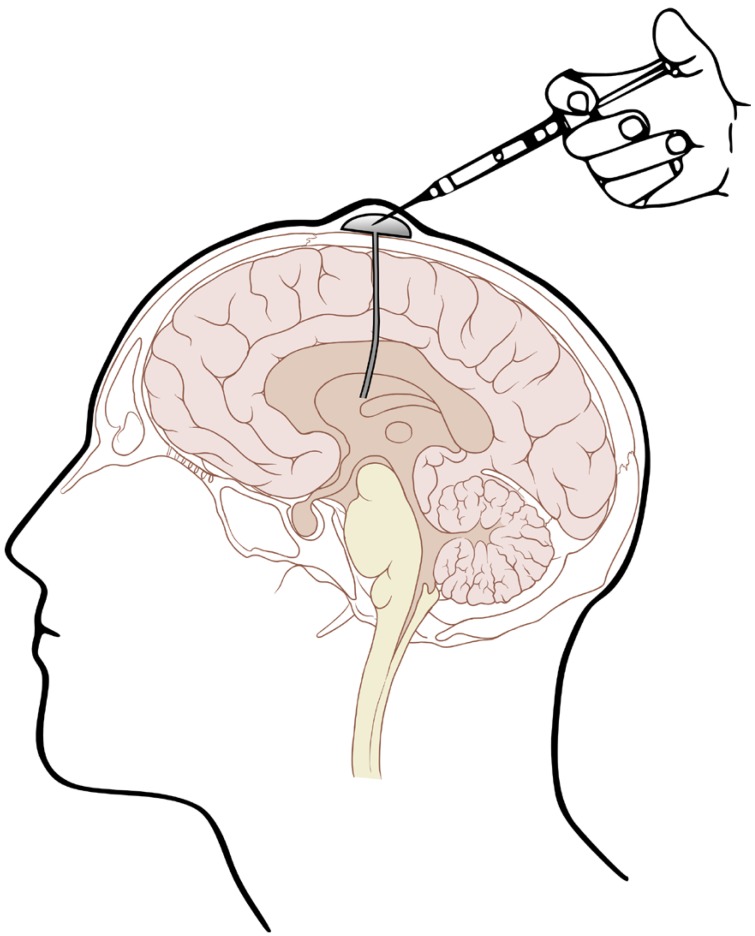
Given the shortcomings of intralumbar administration, Ayub Ommaya, an NIH neurosurgeon, invented a device (Fig. 1) that permits repeated ICV drug administration and CSF sampling.[27] This device facilitated subsequent study of the CSF pharmacokinetics of amphotericin B.[28] This study indicated that amphotericin B exhibits a biphasic distribution pattern in CSF, with the 139 mL distribution volume of the central compartment approximating the expected value for CSF space and a peripheral compartment with a distribution volume of 677 mL, presumably representing brain extracellular fluid space and meningeal drug adsorption. Amphotericin B was eliminated from the CSF at a clearance rate of 0.54 mL/min that was similar to the expected rate of CSF bulk flow through the arachnoid villi. It was found that a daily ICV dose as low as 0.3 mg/day was sufficient to maintain quasi-steady state amphotericin B levels continuously above the minimum inhibitory concentration (MIC) of Cryptococcus neoformans, thus establishing a rational basis for dosing with this mode of therapy.
A subsequent study by Polsky et al.[29] in which patients with cryptococcal meningitis received amphotericin B intravenously either alone or in combination with 0.5 mg/day ICV amphotericin B, demonstrated that the combined therapy was more effective. However, ICV administration of amphotericin B currently is recommended only as salvage therapy for patients in whom intravenous therapy is failing.[30]
Vancomycin
ICV administration of vancomycin has been used successfully to treat meningitis, ventriculitis, and intracranial device infections with gram-positive organisms.[31] Several attempts have been made to characterize vancomycin CSF kinetics but only the report by Pau et al.[32] is technically sound. These authors used vancomycin to treat ventriculitis in a 4-month-old infant with hydrocephalus who had inadvertently received a 45 mg vancomycin dose via a ventriculoperitoneal shunt. CSF concentrations measured over the next week showed the expected biexponential distribution pattern. The following pharmacokinetic parameters were calculated from the authors' results: elimination clearance: 0.03 mL/min, CSF volume: 56 mL, total distribution volume: 131 mL. The value for elimination clearance is consistent with estimates of CSF production rate in neonates[33] and the CSF volume estimate is within the range of 40-60 mL reported for neonates.[34]
Reesor et al.[35] used a one-compartment model to analyze CSF vancomycin concentrations as 3 daily ICV doses of the drug were given to an 82-year-old man with an infected ventriculoperitoneal shunt. They reported that the elimination half-life doubled from 9.3 hr after the first dose to 20.5 hr after the third dose. The authors attributed this increase to “variation” in pharmacokinetic parameters, but their findings are more likely the result of model misspecification. Unfortunately, their data are of insufficient technical quality to permit a more rigorous pharmacokinetic analysis with a more appropriate two-compartment model. A one-compartment model was also used by Hirsch et al.[36] to analyze vancomycin CSF pharmacokinetics in a 25-year-old man with lymphomatous meningitis. However, the calculated values of 60 mL for CSF volume and 0.196 mL/min are unexpectedly low and suggest that tumor blockage interfered with drug distribution and elimination.
As a result of this pharmacokinetic uncertainty, a variety of vancomycin dosing recommendations have been proposed, with single doses ranging from 5 to 60 mg/kg.[37] Therapeutic drug monitoring has been advocated by some authors, with Reesor et al.[35] suggesting that patient-specific pharmacokinetic parameters be calculated from vancomycin CSF concentrations measured after the first dose. These results could then be used to guide subsequent dosing.
Aminoglycosides
Aminoglycosides are another class of antibiotics that do not provide therapeutically effective CSF concentrations when given intramuscularly or intravenously. In a review of treatment options for adult patients with meningitis caused by gram-negative bacteria, the most reliable therapy was found to be gentamicin administered intrathecally in combination with intravenous therapy.[38] In a subsequent study of 6 adolescent and adult patients with gram-negative meningitis, Kaiser and McGee[39] compared intralumbar to ICV administration of tobramycin and gentamicin and reported that distribution throughout the CSF was superior when these drugs were given by the ICV route. Both lumbar and ventricular CSF aminoglycoside concentrations were measured after ICV dosing but a formal pharmacokinetic analysis was not attempted. However, the pooled data that they obtained after a single 5 mg ICV dose can be analyzed with the expected two-compartment model to give the following pharmacokinetic parameters: elimination clearance: 0.25 mL/min, central compartment volume: 143 mL, and peripheral compartment volume: 396 mL. Distribution was complete within the central CSF compartment by 3.5 hours and the gentamicin concentration at that time was 22 µg/mL in both ventricular and lumbar CSF.
Aminoglycosides demonstrate concentration-dependant killing. Given that the minimal bactericidal concentration (MBC) did not exceed 3.13 µg/mL in the study reported by Kaiser and McGee.[39] this gives a ratio of post-mixing maximal aminoglycoside concentration to MBC of 7 that has been shown by Moore et al.[40] to be associated with an 80% likelihood of favorable response in treating systemic infections with gram-negative organisms. The post-antibiotic effect of aminoglycosides also is long enough that the once daily ICV dosing regimen employed by Kaiser and McGee should be suitable when used in combination with intravenous therapy. The general availability of aminoglycoside concentration measurements adds the further possibility of using therapeutic drug monitoring to guide attainment of appropriate peak levels. Trough CSF levels should have no bearing on nephrotoxicity and are of importance only in ensuring that appropriate drug distribution has occurred.[23]
Cytosine Arabinoside (ARA-C) - Pharmacokinetics and a Missed Opportunity
ARA-C pharmacokinetics were first studied by Zimm et al. [41] in seven pediatric patients with acute lymphoblastic leukemia with meningeal metastases. The patients ranged in age from 8 to 14 years. Ommaya reservoirs were used for ARA-C administration and subsequent CSF sampling. ARA-C followed a biphasic pattern of CSF distribution with the central compartment volume averaging 55 mL (range: 32-89 mL). This is less than the average of 90 mL (range: 65-140 mL) reported by Bonadio.[42] for children ranging in age from 4 to 13 years old. Further analysis of the data published by Zimm et al. indicated that the peripheral compartment volume averaged 16 mL. Their reported rate of ARA-C clearance from the CSF averaged 0.42 mL/min, consistent with the expected rate of CSF bulk flow.
Zimm et al.[41] also conducted simulations demonstrating that a thrice daily ARA-C dose of 30 mg would be sufficient to maintain CSF concentrations of this drug continuously above the minimum cytotoxic level for this neoplasm. Because ARA‐C is a cell-cycle phase-specific agent and kills cells only when they are synthesizing DNA, the efficacy of this drug requires continuous maintenance of therapeutic CSF levels. So the inconvenience and increased risk of thrice daily administration has prompted the development of sustained release ARA-C formulations that can maintain therapeutic levels for approximately two weeks after a single dose.[43]
In addition to its use in neoplastic meningitis, ICV ARA-C therapy has also been evaluated in patients with progressive multifocal leukoencephalopathy (PML), a demyelinating disease of the white cortex caused by infection of oligodendrocytes with the JC virus. Padgett and Walker[44] demonstrated that 69% of the 277 adults that they tested had serum antibodies against JC virus and were presumed to have latent JC virus infections. So PML can be regarded as an “opportunistic infection” in that spread of the virus to the central nervous system occurs primarily in latently infected patients whose immune system is compromised as a result of HIV/AIDS, hematological malignancies or, more recently, after institution of therapy with natalizumab and other disease modifying immunosuppressive agents.[45] PML is rapidly progressive, especially in patients with hematological malignancies whose survival averages only 2 years after diagnosis.[35]
In an attempt to treat this disease, the NIH Aids Clinical Trial Group (ACTG) conducted an inappropriately designed multicenter trial in which the unmodified formulation of ARA-C was administered by the ICV route via an Ommaya reservoir to 57 HIV patients with biopsy-confirmed PML.[46] The patients were divided into three groups: one receiving only antiretroviral therapy, a second receiving intravenous ARA-C in addition to antiretroviral therapy, and a third receiving intrathecal ARA-C in addition to antiretroviral therapy. This third group of patients received a 50 mg ICV dose of ARA-C once weekly for four weeks, then once every two weeks for eight weeks, then once every four weeks for the remainder of the 24-week study. The median survival was 15 weeks in the ICV ARA-C group (14 of 19 patients died), 8 weeks in the intravenous ARA-C group (14 of 20 patients died), and 11 weeks in the antiretroviral therapy only group (14 of 18 patients died) (P = 0.85 by log-rank test for group comparison). On the basis of these results, it was concluded that ICV administration of ARA-C is ineffective in treating patients with PML.
In contrast to the elaborate statistical measures that were taken to ensure that the three study groups were appropriately balanced with respect to age, education, disease severity, and CD4+ cell count, the ACTG investigators provided no rationale for the unusual ICV ARA-dose regimen that they selected. Previous in vitro studies had shown that ARA-C in concentrations ≥ 25 µg/mL are needed to completely inhibit replication of the JC virus.[47] So the authors could have used the pharmacokinetic results of Zimm et al.[41] or conducted their own pilot study to design a pharmacokinetically-based regimen that would maintain CSF levels of ARA-C above this concentration. That regimen might have used an extended release formulation of ARA‐C that was available at that time and has a terminal CSF half-life of 5.9 days.[48] So although Hall et al.[46] concluded that ICV ARA-C was ineffective in treating PML, it would appear that the ICV dose regimen that they selected had no chance of being effective. Unfortunately, a properly designed follow-up trial has not been conducted.
Methotrexate (MTX)
MTX is another chemotherapeutic drug that after intravenous administration only attains subtherapeutic CSF concentrations that are only 3%-5% of concurrent plasma concentrations.[49] Bode et al.[49] described the CSF pharmacokinetics of this drug after administering MTX via an Ommaya reservoir to three children whose ages ranged from 8 to 17 years. A biphasic pattern of distribution was observed with a terminal half-life of 6.6 hr. The reported results can be further analyzed to describe a two‐compartment model with a central compartment volume of 47 mL and a peripheral compartment volume of 16 mL. The elimination clearance was 0.27 mL/min. An important facet of this study is that DPTA was injected into the Ommaya reservoir simultaneously with MTX. The clearance of DPTA from the CSF was only 0.22 mL/min, indicating a value of 0.05 mL/min for MTX CSF clearance by presumed active transport. Further evidence for MTX transport from the CSF was obtained when a high dose of probenecid was administered as this prolonged the MTX elimination-phase half-life from 6.6 hr to 7.9 hours, a 19% increment consistent with the above estimate of the contribution of active transport to MTX elimination.This clinical evidence that MTX is actively transported from CSF to blood is consistent with the previous report by Rubin et al.[50] that MTX is transported from CSF at a faster rate than inulin in dogs and that choroid plexus tissue isolated from rabbits can concentrate MTX.
Despite these results, pharmacokinetics has not been used to help design subsequent ICV dose regimens with this drug. Instead, the concentration x time (CxT) concept has been employed to craft rational dosage regimens that maintain MTX concentrations in CSF above the level of 5x10-7 M, considered by Bleyer et al.[21] to be the minimal cytocidal MTX concentration for patients with central nervous system neoplasms. Interestingly enough, the CxT regimen of 1 g intrathecally every 12 hours selected by these authors, did give similar MTX levels in CSF to what can be predicted on the basis of pharmacokinetics. In a further evolution of the CxT approach, Moser et al.[51] administered a daily 2 mg MTX intrathecal dose to patients with recurrent meningeal leukemia and lymphoma for a 3-day course. This course was repeated every 10 days for a total of 4 courses. The authors reported that 14 of the 15 evaluable patients who were treated with this regimen had a complete remission of their recurrent meningeal leukemia or lymphoma.
More recently, Pels et al.[52] treated 20 patients with primary meningeal lymphoma with a 3 mg ICV MTX dose in combination with systemic chemotherapy for 5 consecutive days, followed by an ICV dose of 30 mg of ARA-C to complete a course that was repeated for two more cycles. The authors concluded that the inclusion of ICV therapy in this regimen yielded results that were superior to what they obtained when intravenous chemotherapy was used alone. Although an optimal regimen for ICV MTX therapy is not yet agreed upon, the CxT approach is as effective and less toxic than other regimens in which higher ICV doses have been administered.[21]
Etoposide
Etoposide is a phase-specific cytotoxic drug that interacts with topoisomerase II to inhibit DNA re-ligation and arrest cell replication in the late S phase or early G-2 phase of the cell cycle. Etoposide exhibits dose-regimen dependency and in vitro studies indicate that efficacy increases with duration of exposure but also is dose-limited. By extending the incubation period to 30 hours, Wolff et al.[53] found that maximal cytotoxic effects were obtained with a 0.05 µg/mL concentration of the drug. Henwood and Brogden[54] subsequently reported that in vitro etoposide concentrations of 0.1 to 10 µg/ml were generally cytotoxic, but that cytotoxicity varied with cell line and also was dependent on exposure time.
Fleischhack et al.[55] studied etoposide CSF pharmacokinetics in 4 patients with metastatic brain tumors who ranged in age from 12 to 32 years. They administered a 0.5 mg etoposide dose via an Ommaya or Rickham reservoir and observed a biphasic CSF distribution pattern. Re-analysis of their pooled data using a 2-compartment model gave the following results: central compartment: 67 mL, total distribution volume: 157 mL, elimination clearance: 0.39 mL/min. Although the elimination clearance is consistent with normal values of CSF bulk flow, the small distribution volume suggests that distribution within the CSF space was restricted in these studies, presumably due to tumor blockage. Nonetheless, administration of this etoposide dose for 5 consecutive days maintained trough concentrations above 0.1 µg/mL without accumulation with repeated drug doses, and with clearing of CSF cytology in some of the larger group of 14 patients who were treated with this regimen. These results provided the impetus for a subsequent Phase II study in which ICV etoposide achieved a 26% response rate in a group of 27 patients who received 0.6 mg injected via an Ommaya reservoir for 5 consecutive days, every other week for an 8-week period.[56]
Complications Resulting from ICV Therapy
Implanted devices, such as the Ommaya and Rickham reservoirs that permit direct ICV drug administration, have been in use for over 60 years and their use has generally been safe, even when they have remained in place for several years.[57] However, complications have occurred, primarily as a result of improper ICV catheter placement, toxicity of injected material, or infection. Accidental methotrexate overdoses have caused seizures, coma, and compromised cardiopulmonary function.[58] But glucarpidase (Voraxaze®), a recombinant form of carboxypeptidase G2 that enzymatically cleaves methotrexate, has been used to successfully treat patients with acute MTX toxicity after either intravenous or ICV administration.[59]
Catheter placement
The risks of ICV catheter placement include hemorrhage, postoperative infection, and malpositioning. Whereas an optimally placed catheter tip should be free-floating in a lateral cerebral ventricle, the most common error is placement of the catheter tip too close to the choroid plexus which results in obstructed flow. Initially, after anatomical landmarks were identified, catheters were placed “free hand”. However, in one retrospective report, this technique resulted in accurate catheter placement in only 55% of attempts.[60] Ultrasound and stereotactic guidance were subsequently introduced and these techniques provide nearly 90% accuracy in catheter placement.[60] More recently, frameless electromagnetic image-guidance has been used for ICV catheter placement with even greater convenience and accuracy.[61] These guidance techniques have not only optimized catheter placement and minimized catheter obstruction but have resulted in a lower incidence of procedure-associated intracranial hemorrhage.[61]
Drug selection and formulation
Some drugs, including penicillin and other β-lactam antibiotics with established epileptogenic potential, have caused seizures after ICV administration so are not suitable for intrathecal therapy.[62] Preservative-free drug solutions are preferred for ICV administration and solution pH and osmolarity also must be considered in order to minimize the risk of chemical arachnoiditis.[62] The administered volume should also not be excessive. Rapid mixing within the CSF central compartment will also minimize local toxicity and can be facilitated by depressing the subcutaneously positioned reservoir or Ommaya “pump” (Fig. 1) several times.
Chemical arachnoiditis and leukoencephalopathy
Chemical arachnoiditis or meningitis, following within one day of ICV drug administration and characterized by rapid onset of CSF pleocytosis and symptoms including fever, nausea, vomiting, headache and meningismus occurred in 32% of 60 patients with meningeal malignancies who participated in a Phase II trial of ICV topotecan.[63] However, in a report in which 17 patients with central nervous system embryonal tumors were treated with liposomal ARA-C, these symptoms were minimized by administering prophylactic dexamethazone.[64]
Leukoencephalopathy is a particularly serious complication of ICV therapy. In 5 of 6 patients who had an acute arachnoiditis/meningitis reaction following low-dose ICV methotrexate therapy, a fatal disseminated necrotizing leukoencephalopathy developed 3 to 15 months later.[65] Some, but not all, patients had received whole brain radiotherapy along with ICV methotrexate and this was considered as a possibly contributing factor. In a study of 112 patients with meningeal metastases who were treated with ICV liposomal ARA-C, two patients developed leukoencephalopathy that was centered around the ventricular access catheter.[66] Both patients improved without sequelae after their ventricular access device was removed and they were treated with oral steroids.
Because of the larger volume of post-injection dilution, chemically induced arachnoiditis and meningitis are less likely to occur when drugs are administered by the ICV route rather than by the lumbar route. Similarly, it may be advisable to avoid locally high drug concentrations by confirming CSF patency with DPTA or another suitable marker before using even the ICV route to treat patients with neoplastic meningeal disease.
Nosocomial Infection
Infection remains an important adverse event that occurs either during the surgical insertion of the ICV device or as a result of subsequent improper aseptic reservoir access. In one retrospective study of 616 patients who had Ommaya reservoirs implanted, 34 patients were identified as having infections.[67] Perioperative infections were diagnosed in 32% of these patients, whereas the remainder were attributed to faulty aseptic technique when the reservoir was accessed. Ommaya reservoirs were present in these patients for a median of 316 days and the infection rate was 0.74 infections per 10,000 “Ommaya days”. Coagulase negative staphylococci were the most common infecting organisms.
Concluding Perspective
Despite the lack of large randomized controlled clinical trials to support its use, ICV therapy now has an established place in treating patients with CNS infections and neoplastic disease. Unfortunately, there are only a few rigorous pharmacokinetic studies on which to base rational therapeutic regimens. Even for drugs where pharmacokinetic results are available, for example ARA-C and methotrexate, blockage of CSF distribution pathways poses an additional challenge. It would appear that preliminary imaging or flow studies would be advantageous when ICV therapy is used to treat patients with neoplastic involvement of the central nervous system. Finally, it is hoped that this tutorial will stimulate the interest of clinical pharmacologists in this mode of therapy and will encourage them to conduct the additional pharmacokinetic studies that are needed and to participate in the design of trials that are not pharmacokinetically flawed, such as the AIDS Clinical Trial Group's study of ICV ARA-C in patients with multifocal progressive leukoencephalopathy.[46]
Technical Note
Data for some of the pharmacokinetic analyses was obtained from published figures using the Plot Digitizer program written by Joseph A. Huwaldt (accessed on the Internet at: http://plotdigitizer.sourceforge.net). Pharmacokinetic calculations were made using the SAAM II program (SAAM Institute, University of Washington).
 XML Download
XML Download