

 PDF
PDF ePub
ePub Citation
Citation Print
Print


The kidney is the main target of destruction in chronic kidney disease (CKD). There are currently almost 500 million adults suffering from CKD around the world, and most of them are unaware that they have this disease.[1] In long-term cases of the disease, the kidney cannot filter blood properly and is unable to separate toxins and waste products from the bloodstream. Consequently, these wastes and toxins are accumulated in the blood's circulation and unfortunately accelerate the kidney damage. Dialysis, a renal replacement therapy, which is helpful in discarding these toxins, is often applied to patients in the terminal stage of the disease. However, not all toxins can be eliminated by dialysis. Some toxins that are bound to proteins stay in the circulation.
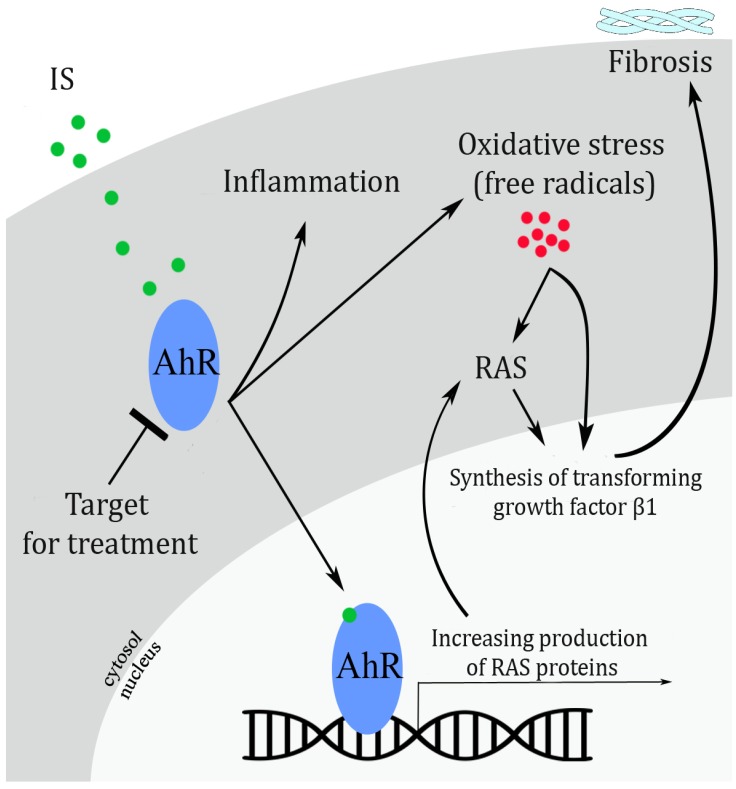
Indoxyl sulfate (IS), as a protein-bound toxin, is recently of interest in the study of CKD. Increased IS in the blood accelerates the progression of CKD in animal studies.[2] Thus, inhibiting the method by which IS causes kidney damage appears to be a promising new strategy for preventing the progression of CKD. This possibility is supported by two online papers published around 3 years ago[34] that reported the mechanism how IS leads to massive kidney damage, and suggested a possible treatment for reducing IS toxicity.
In the first study, Sun et al.[3] investigated the role of IS in activating the local renin-angiotensin system (RAS), widely known to cause heart injury and hypertension. In kidney, this pathway also can cause a hypertension of microvessels and induce cell damage mediated by oxidative stress, inflammation, and fibrosis.[5] They cultured mouse kidney cells for four days in two different media. In the last three days of culture, they added IS to both of the media and added losartan, known as RAS blocker, to the one of the two media. The cells exposed to IS showed not only the increased expression of various proteins involved in the RAS but also transforming growth factor (TGF) β1, known as a fibrosis driver in a variety of organs. On the other hand, the cells exposed to both IS and losartan showed a significant decrease in the expression of the fibrosis, indicating that RAS-targeting drugs can prevent fibrosis development induced by IS.
The authors then investigated the effect of losartan on CKD mouse models and found the same results as in the cultured cell study. Fibrosis and TGF-β1 levels were reduced in mice exposed along with losartan. Showing the link between IS and fibrosis, Sun et al.[3] suggested that the RAS pathway plays an important role in the rapid loss of kidney function and that blocking the pathway results in delayed CKD.
In the second study, Watanabe et al.[4] investigated the effect of IS toxicity on human umbilical cord blood vessels. This study aimed to prove that IS toxicity is caused by the activation of the aryl hydrocarbon receptor (AhR), a protein often involved in herbicide poisoning called agent orange (used as a chemical weapon in 1960's Vietnam War). It is reasonable to suggest that IS activates the AhR, as IS and agent orange are both categorized as toxins. The main difference is that IS is produced within the body whereas agent orange is an external toxin. The mechanism of cell damage by IS was found to be much the same as the mechanism mediated by oxidative stress and inflammation. This study also showed that blood vessels exposed to IS attract white blood cells which contribute to inflammation. Later, Watanabe et al. tested two drugs which block the AhR, and these treatments reduced the effects of IS.
Based on above two studies, IS accelerates kidney damage through the AhR and RAS pathways. However, the exact mechanism has not been known yet. However, some data have highlighted that IS strongly interacts with and activates the AhR (Fig. 1). RAS activity also increases while the AhR is activated. [67] In an experimental study of mice exposed to IS, Ichii et al. [8] showed that the AhR pathway leads to glomerular damage, microalbuminuria, increased the synthesis of mesangial matrix, mesangial cell lysis, tubulointerstitial lesions, vascular damage, and the activation of genes responsible for inflammation. In addition, a cell culture study of podocytes showed that IS caused cell injury, affected the viability of cells, and stimulated the release of cytokines that contribute to inflammation.[8]
Ng et al.[9] also investigated the effect of IS-stimulated AhR activation on RAS-counteracting pathways. The Mas receptor is a protein that counteracts the harmful effect of the RAS. Using human proximal tubular cell cultures, Ng and coworkers revealed that IS reduces the expression of the Mas receptor by activating AhR. From this, we can hypothesize that the interaction between IS and the AhR may precipitate the activation of the RAS pathway, which results in fibrosis and inflammation.
There are some questions following this hypothesis. First, does the pathway described above also affect heart and blood vessels? In fact, the increased level of IS in CKD is related to heart abnormalities and the narrowing and stiffening of blood vesels. Studies of cell cultures and animal models revealed that IS also induces fibrosis and inflammation in the heart and blood vesels. [1011] This indicates that cell damage in the heart and blood vessels is likely to be improved by blocking the AhR.
In addition, the efficacy of AhR-targeting drugs must be examined. Sun et al.[3] showed satisfactory result of using losartan, an RAS-targeting drug, in cells and mice. However, some experts argue that losartan is less effective in blocking the RAS pathway, as some patients with CKD still develop to the terminal stage and die of heart failure even after these drugs are administered. This evidence emphasizes the need for an alternative method of minimizing RAS stimulation in CKD. It is hoped that an AhR blocker can also reduce the expression of the RAS pathway and lead to improved therapies for CKD.
Although these two studies only provide the evidence that IS has effects on CKD development and complications, it is hopeful that providing an effective AhR-targeting treatment may enhance the choice of drugs designed to target the RAS pathway.
 XML Download
XML Download