

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Periodontitis is one of the most common oral diseases in adult populations worldwide and is a major public health concern due to its substantial cost to the medical care system [1]. It is characterized by inflammation and destruction of tooth supporting tissues, in severe cases leading to tooth loss [2]. It is also highly associated with systemic inflammation, resulting in an increased risk for subsequent chronic diseases, such as cardiovascular diseases [3,4], diabetes [5], metabolic syndrome [6-8], pneumonia [9,10], and rheumatoid arthritis [11].
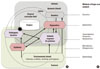
Periodontitis is a complex disease with an etiology involving multiple factors. It includes both extrinsic (modifiable) and intrinsic (nonmodifiable) factors. Although the definitive mechanisms remain unclear, inflammation and infection via outgrowth of multiple opportunistic microbes in the oral environment, including Porphyromonas gingivalis, Tannerella forsythia, and Actinobacillus actinomycetemcomitans, are a contributing factor. [12]. While the presence of microbiological pathogens is a factor leading to this condition, it is not solely sufficient to cause periodontitis. It was recently proposed that dysbiosis of the oral microbiota leads to periodontitis via interference of the host-microbial homeostasis, rather than simple outgrowths of a few pathogens [13,14]. Lifestyle-related factors, such as smoking and dietary patterns, as well as oral hygiene, have also been highly correlated with the prevalence of periodontitis [15,16]. While periodontal research so far has focused on studies of microbiological pathogenesis and oral environments, it is now widely accepted that susceptibility to inflammation is also determined by intrinsic factors such as genetics (Fig. 1A). This is emphasized by the fact that some people are more disease-susceptible or treatment resistant. In this review, we summarize the genetic factors that are of importance in the establishment and progression of periodontitis, and highlight what epigenetic alterations may be critical in the etiology of periodontitis, especially as key mediators between genetic and environmental factors. This information will provide future guidelines for development of novel biomarkers that will aid in the diagnosis, treatment, and cure of this common disease.
PERIODONTITIS-RELATED GENETIC VARIATION
Genetic variations of inflammatory genes
While genetic factors undoubtedly are very important in the development of periodontitis, genetic variations can only become a risk factor when challenged by extrinsic agents and physical insults. Genetic variations linked to complex diseases are not easily identified in multifactorial traits. Single nucleotide polymorphisms (SNP) of the DNA are often used as genetic markers when they can be linked to a distinct phenotype. Per definition, a SNP has to occur in at least 1% of a given population, while individual mutations, not fixed in the population, are referred to as single nucleotide variations (SNVs). Some SNPs alter gene expression levels that may influence host response levels to microbiological growth. For example, SNPs in receptors, antigen sensors in cell surfaces, and cytokines and chemokines have been shown to influence host immunity and inflammatory response [17,18]. Putative periodontitis-related SNPs have been investigated in the Fc-γ receptor (FCGR2A) [19-22], interleukin-1 (IL-1) [23,24], IL-4 [25-27], IL-6 [27,28], IL-10 [29,30], IL-18 [31,32], tumor necrosis factor alpha (TNFA) [23,33,34], vitamin D receptor [21,35-37], cluster of differentiation-14 [38-40], matrix metalloproteinase-1 [41,42], Toll-like receptor-2 (TLR-2) [43,44], TLR-4 [31,32,39,40,43,44], and cyclo-oxygenase-2 (COX-2) [45,46]. These studies suggest a connection between genetic variation and periodontitis at some loci, while their penetrance remains elusive. It is worth emphasizing that the connection between SNPs and periodontitis is not always strong because of variations within populations and subtypes of periodontitis.
Genome-wide analysis of genetic variation and their products
Lately, multiple "omics" technologies have been utilized to increase the understanding of periodontitis disease progression by overcoming the limitations that candidate approaches have provided. The most prominent approaches are genomics, transcriptomics, proteomics, and metabolomics of the host as well as metagenomics of the oral microbiota [47] (Fig. 1B). From a large set of SNPs distributed over all chromosomes, genome-wide association studies (GWAS) have been applied comprehensively to identify genetic variations that are associated with periodontitis [48,49]. GWAS studies have identified novel genes for susceptibility, including GLT6D1 in aggressive periodontitis [48], NIN, NPY, and WNT5A in severe chronic periodontitis, and NCR2 and EMR1 in moderate chronic periodontitis [49]. While it has not been fully determined how the identified susceptibility genes affect pathogenesis, GWAS has provided novel insights into the etiology of periodontitis genome-wide, compared to previously taken candidate approaches.
Furthermore, apart from genome-wide genetic variation analysis, total gene transcript analysis, generally referred to as transcriptomics, has been conducted on periodontal tissues as well as peripheral blood cells [50-52]. Proteomics and metabolomics have also been applied to saliva or gingival crevicular fluid to identify proteins and metabolites with a negative or positive influence on host defense mechanisms [53-55]. It is noteworthy that the levels of transcripts, proteins and metabolites may reflect not only the genetic programming, but also the consequences of response to environmental factors and disease progression. There are layers of chemical modifications on the DNA and its associated proteins that regulate gene expression, commonly referred to as epigenetic effects. These become established or erased based on an environmental response, subsequently leading to intracellular signaling.
EPIGENETICS IN HUMAN DISEASE
Epigenetic modifications
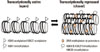
The genetic material, thought of as a database of cellular information, is not only charged by its coding capacity. The last three decades of genetic research has uncovered key factors that reproducibly bind the DNA and organize it into functional units [56]. The DNA is wrapped around histone proteins, the nucleosomes, which serve the purpose of condensing and decondensing the DNA depending on gene activity, and in maintaining chromosome integrity at times of cessation and cell division. The nucleosomes are fairly evenly distributed over the chromosomes, and there are recently developed techniques that allow us to accurately predict where those are positioned [57]. Importantly, each protein in the nucleosome is subject to post-translational modifications, appearing at defined positions of the genome, and is strongly correlated with the activities observed at the DNA (e.g., transcriptional activity and elongation). For instance, trimethylation at histone H3 lysine 4 (H3K4me3) is associated with transcriptional activation, while H3K9me3 and H3K27me3 are associated with transcriptional repression [58] (Fig. 2). The most well characterized epigenetic modification is DNA methylation, which occurs at the 5th carbon of cytosines in mammals (5mC), most commonly next to a guanosine (CpG). Methyl-groups are placed by DNA methyltransferases (DNMTs) that catalyze the transfer from the methyl donor S-adenosyl methionine to the cytosines. Methylation at previously unmethylated sites is placed by the DNMT3a/b, while it is commonly maintained by the DNMT1, referred to as the de novo and maintenance methyltransferases, respectively. Together they assure that the vast majority of the genome is methylated at all times, leaving only regulatory elements like promoters and enhancers, and CpG-rich islands unmethylated [59]. The acquisition of DNA methylation at the promoter is predominantly associated with gene silencing (Fig. 2). DNA methylation can be further modified via oxidation by the TET proteins, implicated in DNA demethylation processes via the base excision repair machinery [60]. On many occasions, multiple epigenetic modifications, including DNA methylation and various histone modifications, work coordinately or antagonistically [59,61,62].
It is easy to conceive that misplaced modifications and the reduced dynamics of their distribution lead to obstructed gene activity and disease (Fig. 3). This generally occurs as a result of mutations of factors that directly make or influence epigenetic modifications [63]. Since each factor has an influence on a genome-wide level, the effect can be dramatic.
Epigenetics in complex diseases
There is an ever increasing amount of evidence showing that understanding the epigenetic pattern in disease progression will provide invaluable information in the diagnosis and treatment of human disease [64]. Genome-wide analysis of epigenetic patterns in tissues that undergo defined changes as a result of external stress have provided insight into how cells respond to external factors [65]. Furthermore, such analysis offers detailed insight into how the cells try to cope with the changes and what may be the outstanding factors that determine whether the changes can be dealt with or result in skewed differentiation or cell death.
The response is hidden in the genetic background, since the results of GWAS have identified correlations with disease susceptibility and progression [66]. This was investigated by screening for quantitative trait loci that are in linkage disequilibrium with genes in close proximity and inferred to be causative in disease predisposition. Extensive studies have been conducted to identify gene expression patterns and/or epigenetically modified loci to determine which ones are correlated with a particular disease [67,68].
An important part of identifying and characterizing the epigenetic pattern in development and disease is to use the information to predict the treatment and cure of diseases based on a suggested genetic response [69]. This information has led to the identification of biomarkers that directly correlate with a defined condition [70]. Similarly, epigenetic patterns that suggest a predisposition for a particular disease have been identified and should be possible to use as the basis for developing personalized and preventive treatment regimes to prevent future problems. However, this is more a vision than a reality in today's medicine.
Epigenetics in environmental response
The placement of epigenetic modifications is tightly controlled both spatially and temporally. Each tissue has a unique epigenetic profile, and changes do occur as a result of developmental and regenerative processes. There is clear evidence that embryonic stem cells have a unique epigenetic pattern that changes upon differentiational cues [71]. Extrinsic factors, such as hormones, regulate differentiation, and in effect influence epigenetic modifications [63]. The epigenetic pattern that we observe in any particular tissue at any particular point in time is a reflection of its activity [72]. Most of the information is then further reflected by its gene expression pattern. Hence, the observed epigenetic pattern can be used to infer the transcriptional condition of the cell or tissue.
Treatment of cells in vitro generates a defined epigenetic pattern, as evidenced by studies of induced pluripotent stem cells [73] and epithelial-to-mesenchymal transition [74]. The gut microbiome can alter the epigenetic pattern of the gut endothelial cells [75,76] by excreting signals that trigger a response. Likewise, a similar effect can be achieved in the oral cavity, which is under the constant influence of extrinsic factors and foreign agents from food intake. Oral hygiene is naturally a contributing factor to oral health. Mounting evidence suggest that a lifestyle of smoking, food intake, lack of exercise, and use of drugs strongly influences the epigenetic pattern and predisposition to most conditions that lead to human disease [77].
EPIGENETICS IN PERIODONTITIS
Inflammation-specific gene expression and epigenetic regulation
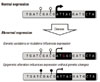
A typical inflammatory response results in the upregulation of genes associated with the production of lectins that then coat epithelial cell surfaces, with the function of recruiting neutrophiles to the site of infection. This initiates an immune response that involves both innate and adaptive-related processes. It is at this stage that epigenetic regulation of gene expression patterns seems to play the most important role [78] and is key in the upregulation of proinflammatory cytokines and other signaling molecules to activate a full response from immune cells, while simultaneously downregulating anti-inflammatory cytokines. The cytokine genes have been suggested as targets of multiple epigenetic events including transcriptional activation via loss of DNA methylation and active histone modifications at regulatory elements [79-81].
The IL-1, IL-2, IL-6, IL-8, IL-10, and IL-12 genes may be regulated by epigenetic mechanisms [79,82]. In chronic obstructive pulmonary disease, proinflammatory cytokines (IL-1, IL-2, IL-8, and IL-12) are highly expressed via increased H3K9 acetylation at the promoters of CBP/p300 and decreased histone deacetylase activity, following the recruitment of NF-kB to gene promoters [83]. TNFA, encoding TNF-α, is also regulated by epigenetic modifications both constitutively and in response to acute stimulation in myeloid cells [84]. DNA methylation also involves cytokine expression such as interferon gamma (IFN)-γ and IL-10 by transcriptional inactivation and skewed differentiation toward IL-10-expressing regulatory T cells, respectively [85,86]. In addition to cytokines, TLR-2 and TLR-4, associated with an increased proinflammatory response, are regulated by DNA methylation in bronchial and intestinal epithelial cells [87,88]. The TLRs, expressed on the cell surface, are involved in the recognition of bacterial components such as lipoproteins, lipo-polysaccharide, flagellin, and DNA [89], so that DNA methylation-mediated regulation of their expression is crucial to determining the magnitude of the bacteria-induced response.
Intriguingly, inflammation signaling itself influences epigenetic changes in cells. IL-6 and IL-1b promote transcription or protein activity of DNMTs, respectively [90,91]. Cytokine-induced methylation changes lead to transcriptional repression of multiple target genes. Taken together, epigenetic mechanisms play a key role in the initiation and progression of inflammation by determination of cytokine profiles in response to environmental stimuli, but also by regulating downstream target genes in response to cytokines.
Epigenetic alterations in periodontitis
Epigenetic studies on the epithelial lining of the oral cavity are in their infancy, but several studies suggest that these cells have a unique capacity to respond to environmental factors. In the periodontal cavity, the inflammatory response involves upregulation of transcription factors (e.g., NF-κB and STAT) and epigenetic chromatin changes similar to other inflammatory diseases [92] (Table 1). Chronic periodontitis patients showed overexpression of cytokines such as IL-6 and IFN-γ in their inflamed tissues [28,93]. The associations between IL-6 and periodontitis are also supported by genetic evidence [27,28]. The expression changes of some loci (e.g., IFNG), occur as a result of the loss of methylation at their promoters [93]. On the other hand, the overexpression of IL-6 is not associated with DNA methylation at its promoter. IL-6 upregulation may rather activate the DNMTs [90], leading to methylation changes at the IL-6-induced target genes and development of a chronic inflammatory condition.
Recently, Zhang et al. [94] showed that the TNFA promoter was hypermethylated at two CpG sites, resulting in decreased expression. By reversing the methylation by treatment with a demethylating agent in vitro, it caused increased expression of TNFA, indicating that the methylation indeed regulated the expression. Lower expression in patients compared to healthy controls was, however, in conflict with a previous report [95]. The authors speculated that the discrepancy might be due to the difference in the state of inflammation of the patients, considering the fact that only severely afflicted patients showed elevated TNF-α [96]. It could also be due to the not-always-direct relationship between the mRNA level and protein level. Either way, further investigations are required to determine the role of TNF-α in periodontitis.
Further evidence of epigenetic changes associated with periodontitis comes from data on COX-2, an enzyme governing the production of prostaglandins that promote inflammation and pain. It has been reported that COX-2 inhibitors were able to reduce the symptoms of periodontitis patients [97]. Nevertheless, COX-2 expression in inflamed gingival tissues from chronic periodontitis patients was lower and its promoter was hypermethylated [98], which was confirmed by an independent study [99]. Similar to TNF-α, methylation changes occur more frequently in periodontitis than in healthy individuals, but it remains unclear whether it is linked to periodontitis etiology or rather indicates the consequence of DNMT activation by persistent chronic inflammation.
In addition to DNA methylation, other epigenetic changes such as histone modifications are involved in periodontitis. Treatment by HDAC inhibitors efficiently suppressed periodontal bone loss in a mouse model of periodontitis [100]. Treatment with novel HDAC inhibitors, such as 1179.4b and MS-275, on P. gingivalis-inoculated mice resulted in significantly reduced bone loss, indicating that maintenance of acetylation is crucial to preventing bone loss.
Collectively, gingival tissues from periodontitis patients seem to have altered epigenetic patterns, particularly at inflammation-related genes. However, it needs to be determined whether 1) the alterations account for the susceptibility like genetic variations in those loci; 2) they are directly related to a mechanism driving the pathogenesis by transcriptional changes in critical target genes; or 3) they are just consequences of chronic inflammatory events. Future genome-wide studies on epigenetic factors promise to provide insightful answers to these questions.
CONCLUSION
The understanding of periodontitis has substantially benefited from the recent identification of genetic factors (in particular SNPs and SNVs) and epigenetic regulatory mechanisms (i.e., aberrant epigenetic patterns). These findings provide novel insight into the etiology of periodontitis, especially regarding the tissue response to infection, as well as highlighting putative mechanisms by which genetic and environmental factors influence each other. While analysis of candidate inflammation-related genetic factors have been common so far, current ongoing genome-wide analysis of genetic variation and epigenetic alterations in periodontitis will likely expand our understanding of the pathogenesis of periodontitis in an unbiased way. The newly gathered information will be used in developing novel therapeutic interventions, potentially involving epigenetic modifiers, leading the way to personalized medicinal treatment and preventional regimes.

 XML Download
XML Download