

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Failure to thrive (FTT) is a physical sign suggestive that a child is receiving inadequate nutrition for optimal growth and development [1]. A wide variety of medical and psychosocial stressors can contribute to FTT, and the work of pediatrician is to determine what may be the leading cause of the malnutrition [1].
When FTT is the result of chronic diarrhea, steatorrhea and the child presents with hypocholesterolemia and fat-soluble vitamins deficit, the diagnosis of a familial hypocholesterolemia (FH) should be keep in mind [2]. Three main genetic disorders classified as FH has been identified: hypobetalipoproteinemia, abetalipoproteinemia and chylomicron retention disease (CRD) [2].
CRD, also known as Anderson's disease, is a rare recessive inherited disorder, being the SAR1B gene, that encodes the Sar1b protein, responsible for this disease [23]. Different mutations as been identified, although it seems that there is no genotype-phenotype correlation [4]. As it was previously reported by Sassolas et al. [5], until 2012, mutations in the SARB1B gene has been established in 43 patients, respective to 17 mutations. Since then, as far as we know, 5 new diagnosis were reported in the literature [6789].
The diagnosis of CRD is often delayed because of their nonspecific signs and symptoms. The first symptoms, which most frequently occur are the gastrointestinal symptoms, namely diarrhea, steatorrhea, vomiting and abdominal distension, resulting in FTT [25]. Hepatomegaly and aminotransferases elevation are also reported in literature, but liver steatosis is infrequent and no instance of cirrhosis has been reported in CRD [25]. Neurological (areflexia, proprioceptive abnormalities, ataxia, tremor, sensory neuropathy), muscular (muscular pain, cramps), ophthalmic (mild defects in color vision and retinal function, micronystagmus) and cardiac manifestations (cardiomyopathy) are described in some older patients with a delayed diagnosis [25]. Probably as a consequence of malabsorption, malnutrition and vitamin D deficiency, these patients may complicate with poor mineralization and delayed bone maturation [2]. Regarding to analytical profile, the presence of hypocholesterolemia with decrease of total cholesterol, high density lipoprotein cholesterol (HDL-c) and low density lipoprotein cholesterol (LDL-c) with normal values of triglycerides, is almost pathognomonic of CRD [2]. The increase value of creatine kinase (CK) may orient toward the diagnosis, however the CK level does not correlate with the severity of neurological manifestations [10]. Other analytical findings are acanthocytosis, hepatic cytolysis, deficiency of fat soluble vitamins and essencial fatty acids (EFA) [2]. The diagnosis is supported by the presence of white duodenal mucosa upon endoscopy, the presence of cytosolic lipid droplets and lipoprotein-sized particles in the enterocytes biopsy and by the identification of a mutation in SAR1B gene [25].
Early diagnosis of this disease is vital, in order to start adequate management with low fat diet supplemented with fat-soluble vitamins, reversing the state of malnutrition and, in this way, preventing clinical progression of the disease with appearance of neurological, cardiac, muscular and ophtalmological signs and symptoms, as previously mentioned [2].
We describe a patient with FTT, chronic diarrhea and steatorrhea who the diagnosis of CRD was established, identifying a mutation never previous described.
Go to : 
CASE REPORT
A 1-month-old girl was admitted in pediatric department because of vomiting and FTT (weight, 3,320 g; −1.97 standard deviation score [SDS]). She was born at 41 weeks, from vaginal delivery, with an adequate anthropometry (weight, 3,130 g [percentile 15–50], length, 49 cm [percentile 50], head circumference, 34 cm [percentile 50]). No complications in the neonate period were identified. Regarding to her family background, there is no inherited or other chronic diseases, but there is a history of consanguinity (first cousin) in her parents. During the hospitalization in the pediatric department, this infant maintained a good appearance and no relevant findings on physical examination were detected. The analytic study, including complete blood count (CBC), kidney and liver function, acid base balance and lipid profile were within the reference values. Urinalysis was normal and urine culture was negative. The radioallergosorbent test for cow milk protein (CMP) was negative and the barium contrast radiography identified gastroesophageal reflux in the lower part of the esophagus, which led to the prescription of proton pump inhibitor and a anti-reflux formula, instead of a standard formula. During hospitalization the vomiting episodes improved, resulting in a weight recovery (28 g/d, weight at discharge of 3,520 g). One month later, she was re-admitted due to worsening of vomiting and weigh lost (weight, 3,440 g; −2.91 SDS). CBC, kidney, thyroid and liver function, lipid profile, cobalamine, folate, iron profile were within the reference values. Urinalysis was normal. Immunodeficiency and metabolic disease were excluded. Thoracic radiography, abdominal and transfontanelar ultrasound didn't identify any relevant finding. At this time, considering the possibility of a non-immunoglobulin E mediated CMP allergy, it was decided to change the anti-reflux formula by a hydrolyzed formula, with a good weight recovery (58 g/d, weight at discharge of 3,850 g).
At 21 month-old this child was referred to the Pediatric Gastroenterology clinic because of feeding refusing, chronic diarrhea, abdominal distension and weight loss. Regurgitation, irritability, hoarseness, abdominal pain, blood or mucus in stools or other gastrointestinal symptoms were denied. Physical examination revealed mucocutaneous pallor, dystrophic aspect and abdominal distension. At this time, her weight was 9,800 g (percentile 10; −1,27 SDS) and her height was 84 cm (percentile 50; 0.01 SDS). The analytical study performed (Table 1) identified iron deficiency anemia (hemoglobin, 11.3 g/dL; ferritin, 8 ng/mL; iron, 16 µg/dL), hypertransaminemia (alanine aminotransferase, 104 UI/L; aspartate aminotransferase, 80 UI/L), fat-soluble vitamins deficiency (vitamin E, 6 µmol/L; vitamin D, 13 ng/mL; prothrombin time, 53%) and hypocholesterolemia (total cholesterol, 52 mg/dL; HDL-c, 13.9 mg/dL; LDL-c, 23.9 mg/dL). Fecal fat quantification confirmed steatorrhea and the essential polyunsaturated fatty acids (PUFAs) were reduced. The remaining study performed excluded coeliac disease, cystic fibrosis, Wilson disease, immunodeficiency, alpha-1 antitrypsin deficiency, auto-immune hepatitis, viral hepatitis and metabolic disease.
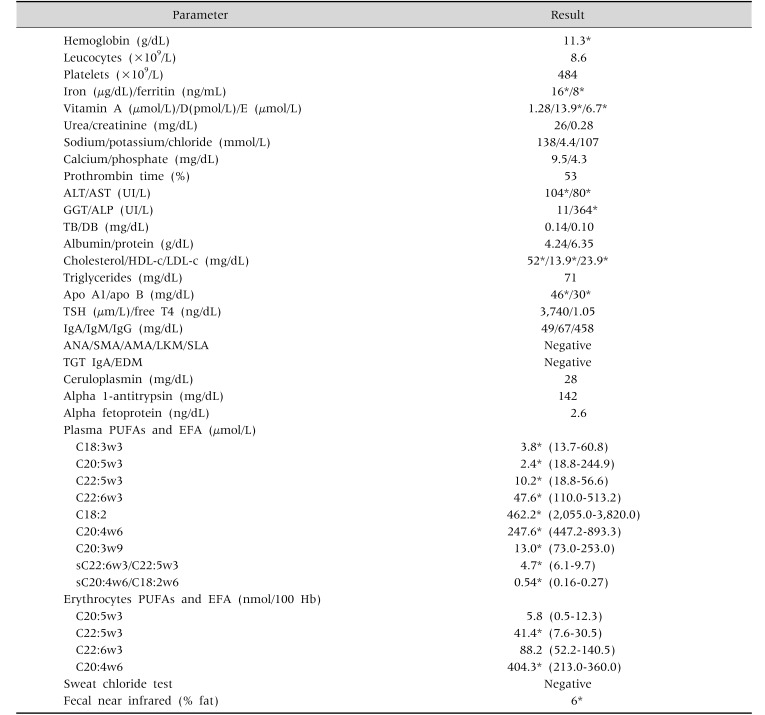
Table 1
Diagnostic Studies Perform at the Time of Referral for Pediatric Gastroenterology Clinic
Diagnostic studies perform at the Pediatric Gastroenterology clinic.
ALT: alanine aminotransferase, AST: aspartate aminotransferase, GGT: gamma glutamyl transferase, ALP: alkaline phosphatase, TB: total bilirubin, DB: direct bilirubin, HDL-c: high density lipoprotein cholesterol, LDL-c: low density lipoprotein cholesterol, TSH: thyroid stimulating hormone, Ig: immunoglobulin, ANA: anti-nuclear antibody, SMA: anti-smooth muscle antibody, AMA: antimitochondrial antibody, LKM: anti-liver kidney microsomal antibody, SLA: anti-soluble liver antigen, TGT: anti-transglutaminase antibody, EDM: anti-endomysium antibody, PUFAs: polyunsaturated fatty acids, EFA: essencial fatty acid.
*Abnormal results.
![]()
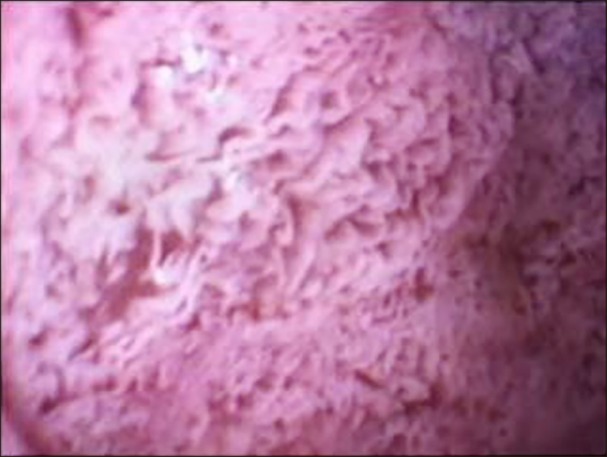
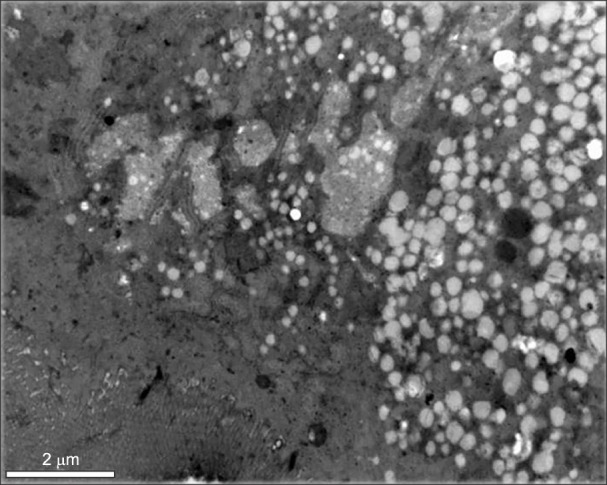
Based upon the clinical and laboratory findings, a preliminary diagnosis of CRD was made and further diagnostic studies were undertaken. The upper endoscopy showed a white coating on the duodenal mucosa (Fig. 1). The intestinal biopsy performed exhibited normal villi, however the enterocytes were overloaded with fat droplets. Electron microscopic examination showed enterocytes with accumulation of lipid droplets (Fig. 2). The SAR1B gene sequencing confirmed a homozigosity mutation in this gene, identifying a mutation never ever described [c.83_84delTG(p.Leu28Argfs*7)]. Since the parents did not intend to have more children, they refused to performed genetic study. However, the mother got pregnant, so she was submitted to a chorionic villi biopsy, which identified the same mutation in heterozygosity, being classified as a healthy carrier. The screening performed in her brothers, confirmed the mutation in heterozygosity in the older boy (8 years old) and excluded affectation in the younger (7 years old).
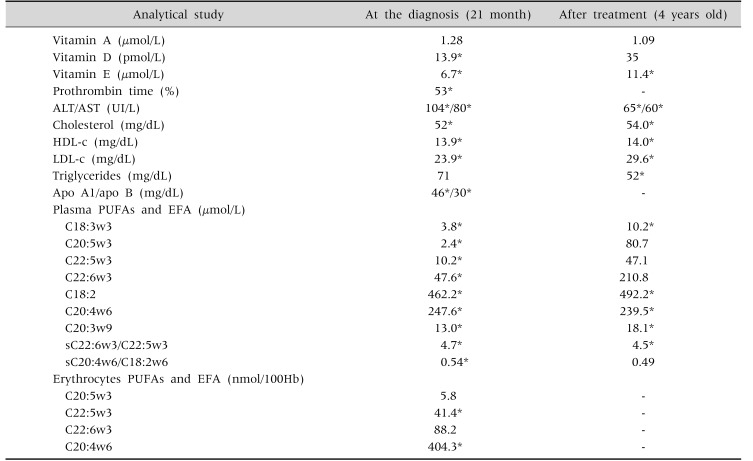
After the diagnosis established this child started a low fat diet supplemented with fat-soluble vitamins, essential fatty acids and medium chain triglycerides (MCT). After this management, a weight recovery, symptoms resolution and analytic improvement was achieved, maintaining hypocholesterolemia and severe vitamin E deficiency, instead of the intramuscular administration and oral high dose of a hydrosoluble form of this vitamin (Table 2). Aiming to identify possible extra-gastrointestinal manifestations, this child was evaluated by ophthalmology, cardiology and neurology that excluded any complication of this disease.
Currently, with 7-year-old, this girl has an adequate anthropometry, a normal cognitive development and she is completely asymptomatic.
Go to :
DISCUSSION
CRD is a rare inherited disease, caused by mutations in SAR1B gene. This gene encodes Sar1b protein, that is responsible for the chylomicron transport from the endoplasmatic reticulum to the golgi apparatus. When this protein is mutated this transport doesn't happen, inducing an accumulation of pre-chylomicrons transport vesicles in the enterocytes [2]. This mechanism seem to be more complex than previous thought, justifying the recent speculations about the existence of mutations in other genes [3].
As shown in this clinical case report, CRD is still a challenge for the pediatrician because of their nonspecific symptoms and the scarce evidence for their management and follow-up [2]. Furthermore, hypocholesterolemia, a common feature of all patients with CRD, may be attributed to malnutrition secondary to chronic diarrhea and the similarity with other FHs, may lead to an incorrect diagnosis [23]. Because all of this particularities, a high index of suspicion and a molecular analysis are necessary to achieve the diagnosis.
In CRD, digestive symptoms are more frequent at the beginning of life. Malabsorptive diarrhea, steatorrhea and abdominal distension are very common and tend to get better within a few days after a low-fat diet is started, as evidence in this clinical case. In fact, after this patient start a low-fat diet supplement with fat-soluble vitamins, EFA and MCT, diarrhea, steatorrhea and abdominal distension resolved and the child regained weight (percentile 50). Tolerance to fat in diet has been reported in a few cases, however in the majority of patients, diarrhea begins again after the reintroduction of fat [5]. Hepatomegaly could be find in 20% of patients, being the discrete hepatic cytolysis a very frequent finding [2]. Although hepatic steatosis is a known complication of hypobetalipoproteinemia, in CRD it was detected in very few cases, and no cases of cirrhosis were reported [25].
Besides the digestive manifestation of this disease, extra-gastrointestinal symptoms can also be present, namely neurologic, ophthalmologic, muscular and cardiac, probably related to impaired Sar1b protein in different systems [24511]. Because of this possibility, our patient was also evaluated by Neurology, Ophtalmology and Cardiology. Neurological examination, assessment of visual acuity and ocular fundus, as well as echocardiogram and electrocardiogram excluded extra-gastrointestinal involvement. This restrictive manifestation of the disease may be due to the early diagnosis established and appropriate and timely management.
Given the nonspecific manifestation of the disease, analytical (hypocholesterolemia with normal triglycerides, deficiency of fat-soluble vitamins and PUFA), imaging (white duodenal mucosa, lipid droplets in enterocytes) and genetic study (SAR1B gene mutation) are necessary for the diagnosis.
Management of these patients is based on a fat free diet, enriched in EFA, MCT and supplementation with fat-soluble vitamins (hydrosoluble vitamin E, 50 UI/kg/d; vitamin A, 15,000 IU/d; vitamin K, 15 mg/wk; vitamin D, 800-1,200 UI/kg/d or 100,000 IU/2 months if younger than 5 year-old and 600,000 IU/2 months if older than 5 years old), as we performed in our patient [2]. In very young children, milk preparations with MCT is necessary to improve diarrhea, although in older children, a regimen of long-chain fatty acids may be sufficient to improve symptoms [2]. Vitamin A and E deficit are implicated in neurological, muscular and ophthalmic complications, so replacement of these vitamins is very important to prevent, slow or improve these kind of complications of CRD [2]. Perhaps due to the timely onset of treatment, no ophthalmic or neurological complications has been detected until now, in our patient.
With this case the authors aim to describe a SAR1B gene mutation never previous described and to remind CRD as a rare cause of FTT, steatorrhea and hypocholesterolemia, highlighting for the importance of an attempting diagnosis, in order to start adequate management, avoiding possible complications.
Go to :
 XML Download
XML Download