

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Lysosomal acid lipase (LAL) is an important enzyme involved in maintaining cholesterol homeostasis inside cells, and it is essential for the intracellular hydrolysis of cholesteryl esters and triglycerides [1]. In the case of lysosomal acid lipase deficiency (LALD), non-hydrolyzed cholesteryl esters and triglycerides accumulate in hepatocytes, resulting in the synthesis of endogenous cholesterol and low-density lipoprotein (LDL). Glycogen storage disease (GSD) is also a metabolic disorder that is caused by deficiency of an enzyme involved in glycogen breakdown, resulting in the accumulation of glycogen in hepatocytes.
Both diseases present a very similar clinical manifestation and histologic findings that may lead to misdiagnosis. Therefore, careful differential diagnosis between LALD and GSD is important. Here, we report the first Korean patient with LALD who was previously misdiagnosed with GSD.
CASE REPORT
A 6-year-old boy was referred with abdominal distension. The patient was sent to an orphanage when he was 1 year old; therefore, his specific birth history is unknown. At the age of 2, he had a seizure but did not receive any evaluation. When he was 3 years old, he was brought to a hospital because of abdominal distension, poor weight gain and developmental delay. A blood test showed elevated liver enzymes, and hepatomegaly was observed by abdominal ultrasonography. Liver biopsy revealed swollen hepatocytes with flocculent cytoplasm and septal fibrosis. He was diagnosed with GSD. At 6 years of age, he was transferred to Seoul National University Children's Hospital for further diagnostic evaluation and proper management.
At this time, his height was 107.3 cm (<3rd percentile), body weight was 16.9 kg (<3rd percentile) and head circumference was 49 cm (5–10th percentile). His blood pressure was 123/77 mmHg (>95th percentile), pulse rate was 102/min, respiratory rate was 24/min, and body temperature was 37.1℃. His abdomen was soft but distended. There was no tenderness or rebound tenderness. The liver was palpated four finger breadth below the costal margin. The spleen was palpated two finger breadth below the costal margin.
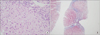
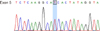
Liver magnetic resonance imaging (MRI) revealed a hepatic fat percentage of 6.35%. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels were elevated at 72 and 76 U/L, respectively. His lipid profile was also elevated, with a total cholesterol of 226 mg/dL and an LDL of 192 mg/dL. His high-density lipoprotein (HDL) level revealed a moderate reduction of 19 mg/dL. The patient's serum calcium level was slightly elevated at 11.9 mg/dL; however, the parathyroid hormone level was normal (8 pg/mL) . The findings of the liver biopsy, through light microscopy, showed diffuse microvesicular fatty changes in hepatocytes and septal fibrosis (Fig. 1). The portal spaces also showed some foamy macrophages. Ultrastructural examination revealed that the cytoplasm of the liver cells contained numerous lysosomes filled with a lipid material and intracytoplasmic cholesterol clefts, suggesting lipid metabolic disorder (Fig. 2). The patient's LAL activity, determined by a dried blood spot test, was 10.2 pmol/punch/h, which was considerably lower than the normal value (controls, 265.4±78.3; carriers, 107.1±30.7). Molecular genetic analysis of the LIPA gene was performed for confirmative diagnosis. Direct sequencing analysis of LIPA identified a novel homozygous mutation, c530C>T (p.Thr177Ile), in exon 5 (Fig. 3). The Polyphen-2, Sorting intolerant from tolerant and Mutation Taster programs predicted p.Thr177Ile to function as a pathogenic mutation. Two siblings of the patient were evaluated pre-symptomatically. Genetic analysis of the two siblings showed that one is a heterozygous carrier, while the other is unaffected.
Enzyme replacement therapy (sebelipase alfa 1 mg/kg every 2 weeks) was initiated, and the patient consequently showed improvements in transaminases and the lipid profile as well as a reduction in liver volume.
Marked decreases from baseline in AST (72 U/L at baseline, decreased to 44 U/L), ALT (76 U/L at baseline, decreased to 40 U/L), and LDL (192 mg/dL at baseline, decreased to 134 mg/dL) were seen after 10 months of treatment. In addition to the improvements in transaminases and the lipid profile, liver MRI revealed significant reduction in hepatic fat percentage (6.35% at baseline, decreased to 3.06%).
DISCUSSION
LALD is a disorder caused by deficiency in LAL activity, resulting in the accumulation of cholesteryl esters. Nearly all LALD patients present with hepatosplenomegaly and growth retardation [2], and most patients show elevated hepatic transaminase (AST/ALT), serum total cholesterol and LDL levels, while HDL levels are decreased [3].
In the present case, the patient had been previously diagnosed with GSD. GSD is an inherited disorder of carbohydrate metabolism that is caused by deficiency in an enzyme involved in glycogen breakdown, resulting in the accumulation of glycogen in hepatocytes. Similar to LALD patients, hepatosplenomegaly, dyslipidemia, and growth retardation are the main findings in GSD [45]. However, unlike LALD patients, GSD patients may present with fasting hypoglycemia [6].
For the differential diagnosis of LALD and GSD, the findings of a liver biopsy are important.
Liver biopsy specimens collected from the current patient revealed diffuse microvesicular fatty changes in hepatocytes and septal fibrosis; the portal spaces also showed some foamy macrophages. Ultrastructural examination demonstrated that the liver cell cytoplasm contained numerous lysosomes filled with lipid material and intracytoplasmic cholesterol clefts. GSD patients also showed pale cytoplasmic changes in hepatocytes and occasional microvesicular steatosis in liver biopsies [3]. However, prominent nuclear hyperglycogenation and swollen hepatocyte are the key diagnostic features of GSD [7]. Additionally, the liver typically contains particles that are strongly stained with Periodic Acid-Schiff and can be decolorized using a bleaching agent; in addition, septal fibrosis is generally detected in GSD type 3. In contrast to GSD, periportal foamy macrophage and intracytoplasmic cholesterol clefts are the main diagnostic features of LALD. Because of their overlapping clinical and histological findings, careful differential diagnosis is important.
The current patient had hypertension upon physical examination when he was initially evaluated. The cardiovascular manifestations of LALD patients include accelerated atherosclerosis, heart failure, aortic calcifications, myocardial infarction, and stroke. Hypertension was previously reported in 3 patients between 4 and 8 years of age [3].
The current patient's serum calcium level was slightly elevated to 11.9 mg/dL, although an evaluation of hypercalcemia revealed no abnormality. There have been no reports of hypercalcemia in LALD patients.
To date, over 40 LIPA mutations causing LALD have been identified. The most common LIPA gene mutation in Caucasians is an exon 8 splice-junction mutation (E8SJM-1G>A), which results in altered mRNA splicing and exon 8 skipping [3]. However, the LIPA gene mutations reported in Japanese patients include Tyr22X, Val203 Leu and Leu264Pro [89]. Additionally, sequencing of the LIPA gene in this Korean patient revealed a novel homozygous mutation in exon 5 (p.Thr177Ile). These findings in Korean and Japanese patients suggest that LIPA gene mutations in Asians maybe different from those in Caucasians. Consequently, estimation of the prevalence of LALD in Asian populations requires further study, as E8SJM-1G>A is not common in these racial groups [10].
Sebelipase alfa is a recently developed treatment that can modify the natural course of the illness. As shown in this case, sebelipase alfa produced a rapid decreases in serum transaminases, improvements in the serum lipid profile and a reduction in liver and spleen size [1112].
In summary, we report the first Korean case of LALD with a novel LIPA mutation. This patient had been previously misdiagnosed with GSD. Although rare, LALD should be considered in the differential diagnosis of a child with hepatosplenomegaly and dyslipidemia.

 XML Download
XML Download