

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Carnitine palmitoyltransferase 1A (CPT1A) is a hepatic isoform of CPT1, which is the enzyme located in the outer mitochondrial membrane and is important for mitochondrial fatty acid oxidation (FAO) [1]. Without CPT1A, long chain fatty acids cannot be transported into mitochondria to be broken down, processed and stored in our body. CPT1A deficiency (OMIM#255120) is a rare metabolic disease with impaired mitochondrial FAO and results in accumulation of high levels of long chain fatty acids in the liver. Therefore, patients with CPT1A deficiency usually present hypoketotic hypoglycemia and hepatic encephalopathy after long time of fasting.
CPT1A deficiency is caused by mutation in the gene encoding CPT1A, the CPT1A gene. The prevalence appears to be quite low worldwide and fewer than 40 cases are reported to date, though c.1436C>T (p.Pro479Leu) variant of CPT1A was known to be prevalent in Inuit population [2]. Just a single case in Korea with CPT1A deficiency was reported in 2015, and demonstrable hepatosplenomegaly and nephromegaly in the patient were mentioned as atypical manifestations of CPT1A deficiency [3].
In the present report, we describe a case of a Korean boy with CPT1A deficiency showing cholestatic jaundice as the first manifestation of CPT1A deficiency.
CASE REPORT
A 1.9 years old boy was referred to the emergency room in Seoul National University Hospital (Seoul, Korea) to evaluate and manage jaundice developed after viral gastroenteritis. The boy was born at full term with 2.97 kg (10-25th percentile) of birth weight by cesarean section, and was the only child of nonconsanguineous and healthy parents. The result of his neonatal screening test for inherited metabolic diseases performed at the age of 3 days was normal, and he had been normally developed. Ten days before the visit to our hospital, he was admitted for treatment of respiratory syncytial virus infection and noroviral enteritis at a local hospital. Icteric sclera was first observed after a week of the admission, and hyperbilirubinemia along with elevated levels of liver enzyme were noticed and had been progressed in repeated tests before the referral to our hospital.
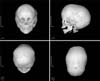
During examination at our hospital, jaundice appeared on his face and abdomen, icteric sclera and scaphocephaly were observed. The height and weight of the boy were 85 cm (25-50th percentile) and 10.7 kg (3-5th percentile), respectively and his head circumference was 49.7 cm (50-75th percentile). He could walk alone and walk up and down the staircase. Moreover, he could speak many meaningful words. The liver was palpable 4 cm below the right costal margin. The findings of initial laboratory examination performed in our hospital, revealed elevated levels of bilirubin and liver enzymes: total bilirubin, 8.0 mg/dL (normal 0.2-1.2 mg/dL); direct bilirubin, 5.7 mg/dL (normal 0-0.5 mg/dL); aspartate aminotransferase (AST), 234 IU/L (normal 1-40 IU/L); alanine transaminase (ALT), 168 IU/L (normal 1-40 IU/L); and alkaline phosphatase, 480 IU/L (normal 60-300 IU/L). Viral hepatitis serology markers including hepatitis A, B, and C viruses, cytomegalovirus, and Epstein-Barr virus were not informative with respect to diagnosis. Prothrombin time and activated partial thromboplastin time) were 13.6 sec (normal 9.6-12.5 sec) and 48.1 sec (normal 26-35.3 sec), respectively. Serum ammonia level was 89 µkg/dL (normal <75 µg/dL). Hemoglobin was 8.2 g/dL (normal 10.5-14.0 g/dL) and iron deficiency (iron 53 µg/dL, normal 50-170 µg/dL; total iron-binding capacity 479 µkg/dL, normal 280-400 µg/dL; ferritin 8.8 ng/mL, normal 10-300 ng/mL) was noted with no definite evidence of hemolysis. Ceruloplasmin, creatine phosphokinase, and lactate dehydrogenase levels were normal. Compensated metabolic acidosis with normal anion gap (pH 7.38; HCO3-, 18.3 mmol/L; Na, 136 mmol/L; Cl- 103 mmol/L) was found on blood gas and electrolyte analyses, and the presence of renal tubular acidosis (RTA) was suggested. Tubular dysfunctions including mild tubular proteinuria were observed (spot urine ratio of beta-2 microglobulin/creatinine 1.52 µg/mg, normal 0.09-0.48 µg/mg; N-acetyl-beta-glucosaminidase/creatinine 206 IU/g, normal 2.9-20.0 IU/g; protein/creatinine 0.23 mg/mg, normal <0.3 mg/mg). Abdominal ultrasonography revealed hepatomegaly with coarse parenchymal echogenicity without focal lesion or bile duct dilatation. Enlarged kidneys with increased renal cortical echogenicity were also observed (Fig. 1). Skull simple radiography and three-dimensional computed tomography images showed premature closure with sclerotic change of the sagittal suture, suggesting craniosynostosis (Fig. 2).
Additionally, we performed tandem mass spectrometry (TMS) analysis, carnitine assay, serum amino acid and urine organic acid analyses for screening of metabolic liver diseases. The results of TMS and carnitine analyses showed elevated levels of free and total carnitine to 77.6 µmol/L (normal 18.0-54.0 µmol/L) and 87.5 µmol/L (normal 26.0-55.0 µmol/L), respectively; however, acylcarnitine level was normal at 9.9 µmol/L (normal 4.0-19.7 µmol/L). On suspicion of CPA1A deficiency, carnitine analysis was repeated after 2 weeks later and low fat and high carbohydrate diet with medium chain triglyceride oil and iron supplementation was started concurrently. The result of repeated carnitine analysis was reported a week later and revealed further elevated levels of free and total carnitine to 101.1 µmol/L and 104.3 µmol/L, respectively and a decrease in acylcarnitine value to below the normal range (3.2 µmol/L), therefore based on the laboratory reports, CPT1A deficiency could be suspected. Molecular genetic analysis of the CPT1A gene was performed for confirmative diagnosis. Direct sequencing analysis of CPT1A led to identification of two novel mutations, c.1163+1 G>A and c.1393G>A (p.Gly465Arg) (Fig. 3), and the patient was diagnosed to be suffering from CPT1A deficiency.
With diet management, hyperbilirubinemia and elevated liver enzymes were progressively improved (total bilirubin, 1.5 mg/dL; AST, 37 IU/L; ALT 22 IU/L) (Fig. 4). One month after diagnosis, cranioplasty for scaphocephaly was performed. During the post-operation care, recurrent hypoglycemic events with desaturation and tachycardia occurred even though glucose solution was continuously infused during that time (glucose infusion rate, 3.3 mg/kg/min). Recovery of vital signs and mental status was achieved after increased glucose infusion rate with 10% dextrose solution (glucose infusion rate, 5.5 mg/kg/min).
At present, the boy is 2.3 years old, laboratory findings including liver enzymes and bilirubin have become normalized (AST, 35 IU/L; ALT, 30 IU/L; total bilirubin, 0.3 mg/dL; direct bilirubin, 0.11 mg/dL). Hemoglobin and RTA features have also been improved (hemoglobin, 10.9 g/dL; pH 7.39; HCO3-, 21.8 mmol/L; Na, 134 mmol/L; Cl-, 110 mmol/L). Although hepatomegaly and nephromegaly were still observed on the follow-up ultrasonography, he has shown catch-up in weight (12.1 kg, 10-25th percentile) and exhibit normal developmental milestones.
DISCUSSION
CPT1A deficiency is a rare metabolic disease in FAO, and the majority of patients have demonstrated recurrent hypoglycemic events as the first manifestation [4]. Occurrence of hypoglycemia has been noted when glycogen was exhausted often with long time of fasting or infection. However, in the present case, the patient did not demonstrate any hypoglycemic events before diagnosis. There exists a report of a patient with cholestatic jaundice along with CPT1A deficiency [5], in which the authors have described existence of cholestatic jaundice due to impaired energy preventing adenosine triphosphate (ATP)-dependent bile acid secretion. In addition, Greenberg et al. [6] suggested that homozygous subject for a certain mutation, c.1436C>T (p.Pro479Leu), could be asymptomatic even though it caused the low residual CPT1A activity. It is also suggested that milder forms of CPT1A deficiency could be sufficient to supply glucose through mitochondrial FAO, since these are insufficient in ATP-dependent bile acid secretion system. In our patient, it is hypothesized that systemic hypoglycemia might not be definitely accompanied and jaundice occurred as the first manifestation of CPT1A deficiency instead. Early diagnosis is usually difficult before progression of hepatic dysfunction in these patients. Therefore, screening for metabolic disorders including TMS analysis, carnitine assay, serum amino acid and urine organic acid analyses, should be considered as the initial evaluations for patients showing unusual course of jaundice.
TMS analysis on dried blood spots can detect more than 20 metabolic disease including CPT1A deficiency [7]. A Japanese report states detection of CPT1A deficiency in a presymptomatic baby by TMS newborn screening [8]. The patient did not show hypotonia, hypoglycemia, RTA and jaundice. However, elevation of free carnitine level was noticed by TMS newborn screening. Although our patient was not detected by newborn screening using TMS in the neonatal period. TMS usually uses some indicators including C0 level, C16 level, C18 level, or C0/(C16+C18) ratio to diagnose CPT 1A deficiency. Fingerhut et al. [7] reported 2 false negative cases in the method using C16 level, and C0 / (C16+C18) ratio is thought to be a very specific indicator, but can be normal even in patinets [2]. Our patient was not a severe neonatal onset form of CPT1A deficiency, and the normal result in his neonatal TMS was attributed to his milder clinical severity. So repeated TMS analyses were useful and necessary to detect CPT1A deficiency for suspected patients.
Molecular genetic analysis for the CPT1A gene is the confirmative test for diagnosis, and approximately 30 kinds of mutations have been reported to date (the Human Gene Mutation Database, http://www.hgmd.cf.ac.uk). There are well-known founder mutations, c.1436C>T (p.Pro479Leu) in Inuit population and c.2129G>A (p.Gly710Glu) in Hutterite populations has been reported in previous studies [1910]. Targeted mutations analysis can be accepted for the known mutations in these ethnic groups. However, direct sequencing analysis for all coding exons was usually used in other ethnic groups, who have low prevalence of CPT1A deficiency as Koreans. We performed direct sequencing analysis of all coding exons and identified two novel mutations, c.1163+1G>A and c.1393G>A (p.Gly465Arg). Although family screening of these mutations for patient's parents was not performed yet, each of these two mutations is thought to be pathologic mutation. The c.1163+1G>A is located at the spicing donor site of intron 10 and cause exon 10 skipping and c.1393G>A (p.Gly465Arg) is located on exon 12. The 465 glycine is a highly conserved amino acid throughout species, and another mutation at the same amino acid site, p.Gly465Trp, was already reported in the CPT1 patient [11]. Genetic counseling through family screening of identified mutations should be necessary, if the parents want to have another baby.
RTA and nephromegaly were also observed in our patient. RTA in CPA1A deficient patients have been known from the previous reports [12]. The kidney is rich in mitochondria, and fatty acids are thought to be an important source of energy in the kidney. Therefore, RTA can be an evidence of lack of energy for the kidney with impaired fatty acid metabolism and can be resolved following treatment with medium chain triglyceride. Severe nephromegaly with significant proteinuria were also reported in another study [3]. Accumulation of long chain fatty acid and dyslipidemic changes in kidney may lead to nephromegaly in CPT1A deficient patients. During examination of tubular functions performed in our patient, mild tubulopathy was usually observed, but there were no other significant abnormal findings including proteinuria and hematuria on urine analysis.
A sufficient level of glucose should be supplied to patients with CPT1A deficiency during long time of fasting. Our patient showed hypoglycemia and hepatic encephalopathy under stressful situations, even though sufficient glucose (3.3 mg/kg/min) was continuously infused during fasting after surgery. It suggests that higher glucose infusion rates should be kept in CPT1A deficient patients, and frequent blood glucose level monitoring is also necessary during stressful conditions including major operation and severe infection. Blood glucose level had been normalized with higher glucose infusion rate (5.5 mg/kg/min) in our patient. Before the diagnosis, our patient did not show symptoms such as hypoglycemia and encephalopathy, and his usual appetite and nutrition intake seemed not to be extraordinary. However, he showed difficulty in gaining weight, which indirectly represents his undetected poor nutritional status.
Craniosynostosis has not been reported as the manifestation or comorbidity of CPT1A deficiency. Our patient showed sagittal craniosynostosis and it is the most common subtype of non-syndromic craniosynostosis, which is not associated with strong genetic backgrounds. Therefore, it is hypothesized that adjunction of sagittal craniosynostosis in our patient might be unrelated to CPT1A deficiency.
In the present work, we report a patient with CPT1A deficiency, who showed cholestatic jaundice as the first manifestation rather than hypoglycemia. Elevated free and total carnitine levels and decreased acylcarnitine level were the clues for clinical diagnosis. Also, two novel mutations of CPT1A, c.1163+1G>A and c.1393G>A (p.Gly465Arg), were identified. Accordingly, FAO including CPT1A deficiency should be considered to be a rare cause of cholestatic hepatitis in childhood.



 XML Download
XML Download