

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Conjugated hyperbilirubinemia signifies cholestasis from underlying hepatobiliary dysfunction. This finding at any age is abnormal. Cholestasis results from impairment in the excretion of bile and its constituents secondary to defective transmembrane transport or mechanical obstruction. Histological findings in cholestasis show evidence of bile pigments in hepatocytes. Clinically there is also accumulation of bilirubin, bile acids, and cholesterol in the blood and extrahepatic tissues. This accumulation results in the clinical findings of jaundice, scleral icterus, xanthomas, and cholestatic pruritus [1]. The mechanism of cholestatic pruritus is multifactorial and linked to increased itch causing molecules hy-pothesized to be bile salts, endogenous µ-opioids, his-tamine, serotonin, and steroids [23]. Cholestatic side effects, especially significant pruritus, can negatively affect patients' quality of life by disrupting daily activities and sleep [4]. Further diagnostic testing should never be delayed.
One sign of cholestasis is the manifestation of jaundice which refers to the yellow discoloration of the skin, sclera, mucous membranes, and body fluids. Jaundice is apparent when serum total bilirubin exceeds 2 to 3 mg/dL (34.2 to 51.3 µmol/L) in older children. Cholestatic hyperbilirubinemia is defined as serum conjugated bilirubin greater than 1.0 mg/dL (17.1 µmol/L) if the total bilirubin is less than 5.0 mg/dL (85.5 µmol/L), or greater than 20% of the total serum bilirubin if the total serum bilirubin is greater than 5.0 mg/dL (85.5 µmol/L) [5]. The pathophysiology of jaundice is complex and is determined by the specific etiology. However, there are several common features found in chronic cholestasis including clinical presentation, symptoms, and nutritional management.
PATHOPHYSIOLOGY
Cholestasis results from impairment in the excretion of bile secondary to mechanical obstruction of bile flow or altered excretion of bile components into the bile canaliculus. Bile has many constituents including bile acids, phospholipids, cholesterol, bilirubin, heavy metals, as well as a number of metabolites [67]. Bile formation and transport is finely regulated and dependent on the function of distinct membrane transport systems [89]. Genetic mutations in transporter genes or exposure to cholestatic injury result in reduced expression and function. This results in impairment in the metabolism and excretion of any of these constituents. This, in turn, leads to cholestasis.
One element of bile, conjugated bilirubin, is a diagnostic indicator of cholestasis. If any of the components of bile cannot be transported into the canaliculus, or if there is mechanical obstruction in the bile ducts, the bilirubin re-circulates into the blood stream and levels are elevated in the serum. Bilirubin is a red flag found in the laboratory evaluation that can help the clinician identify cholestasis. Thus, understanding the metabolism of conjugated bilirubin is essential for deconstructing the complex mechanisms of cholestasis.
Unconjugated bilirubin, a precursor to conjugated bilirubin, is poorly water soluble, bound to albumin in the circulation, and a product of heme breakdown. Bilirubin is dissociated from albumin and taken up by hepatocytes via the carrier bili-translocase at the sinusoidal junction.
Within the hepatocyte, unconjugated bilirubin is bound to glutathione s-transferase. It is then conjugated with glucuronic acid via bilirubin glucuronosyltransferase. Conjugated bilirubin is excreted into bile canaliculi via an adenosine triphosphate dependent transporter known as multispecific organic anion transporter or multidrug resistantce-associated protein (MRP2). If there is any interruption of transport of bilirubin into the canaliculus, the conjugated bilirubin will recirculate into the serum, where it is detected on laboratory evaluation.
FOCUSED DIFFERENTIAL OVERVIEW
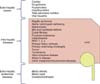
Beyond the neonatal period, the differential for cholestasis is broad and depends on the clinical presentation, physical exam findings and the chronicity of the disease. This review article focuses on only a few key causes of cholestasis. Other important causes such as un-operated biliary atresia, failed Kasai procedures, mitochondrial diseases, or cholestasis secondary to herb ingestion are important causes of cholestasis but are not discussed in detail in this article as we have only selected a few etiologies to discuss in depth. Fig. 1 provides a more comprehensive differential diagnoses for cholestasis among children and adolescent anatomically.
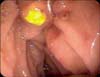
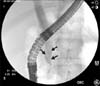
The most common cause of biliary obstruction in older children is cholelithiasis. Choledocholithiasis, the presence of one or more gallstones lodged in the common bile duct, is a common cause of cholestasis and conjugated hyperbilirubinemia. One study reviewed a single center's experience with pediatric gallstones and reported that 72% of the stones were pigmented, 17% were cholesterol stones, and 11% were of unknown composition [10]. While the cause and associated conditions vary among different age groups within the pediatric population, total parenteral nutrition administration, abdominal surgery, hemolytic disease, and hepatobiliary disease were common causes across age groups from birth until 11 years of age [11]. Although no specific gene has been linked to symptomatic gallstone disease in the pediatric population, rates of disease prevalence among twin studies revealed an increased concordance rate among monozygotic twins [12]. Cholestasis caused by choledocholithiasis is often transient and may resolve spontaneously with stone passage or with the help of therapeutic endoscopic retrograde cholangiopancreatography (ERCP) (Fig. 2 and 3).
Biliary cysts, previously described as choledochal cysts, can also present with cholestasis. Biliary cysts are cystic dilations that may occur throughout the biliary tree. They are divided into five different types depending on the location, number of cystic dilations, and involvement of the pancreaticobiliary junction (Table 1). Incidence of biliary cysts has been reported at up to 1 in 1,000 births in select Asian countries, and is higher among Europeans and North Americans [13]. Many mechanisms have been proposed for the pathogenesis of congenital and acquired biliary cysts. The majority of patients present before the age of 10 years of age, usually with a triad of abdominal pain, jaundice, and a palpable mass [1415]. Cysts can also be incidental findings on abdominal imaging, including ultrasound or computed tomography (CT) scans. Biliary cysts are associated with a number of complications, including cholelithiasis, ductal strictures, cholangitis, and even cholangiocarcinoma. These complications present with a cholestatic pattern of serum liver tests.
Wilson's disease is an autosomal recessive copper storage disease linked to mutations on the ATP7B gene [16]. The carrier rate of Wilson's disease was previously described as 1 in 90 worldwide, with a prevalence of approximately 1 in 30,000 [1617]. A recent study evaluating the prevalence of the disease after the discovery of the ATP7B gene linked to Wilson's indicates that the incidence is even higher than previously described [18]. The disease hallmark is decreased incorporation of copper into ceruloplasmin and resultant decrease of copper excretion in the bile. As a result, copper accumulates principally in the liver and brain, causing progressive hepatic and neuropsychiatric disturbances [19]. While copper accumulation starts at birth, symptoms rarely present before five years of age. Approximately, 40-60% of patients present with symptoms during the second decade of life [1]. Symptoms are often non-specific and will require a high degree of suspicion by the clinician. In the pediatric population, a patient will present more often with hepatic abnormalities because the central nervous system appears to be affected at a later time period [19].
Progressive familial hepatocellular cholestasis (PFIC) is a collection of inherited disorders that lead to progressive liver failure secondary to impaired bile formation and secretion. This conglomerate of diseases often presents in infancy and childhood with intractable pruritus, coagulopathy, and other signs and symptoms of end stage liver disease. While prevalence is unknown, the estimated incidence is about 1 in 50,000 to 1 in 100,000 births [20]. Three broad types of PFIC have been described with an autosomal recessive mode of inheritance [21]. Type I presents with unremitting cholestasis, pruritus, and jaundice prior to one year of age [202122]. Of note, serum gamma glutamyltransferase (GGT) level is typically normal in PFIC type I, normal in type II, and elevated in type III [2122]. MDR3 gene abnormality is seen in PFIC type III, which leads to abnormal secretion of phospholipids. The bile acids therefore accumulate and act as detergents to remove the GGT protein from the connecting glycosyl phosphatidyl inositol that anchors it to the bile canalicular membrane. The GGT protein then circulates into the serum and is elevated on laboratory in patients with PFIC III. Patients with PFIC often progress to cirrhosis and resultant liver failure.
Alagille syndrome is an autosomal dominant disorder characterized by genetic abnormalities of JAG1 and the Notch signaling pathway. It is histologically characterized by paucity of intrahepatic bile ducts leading to chronic cholestasis [23]. In addition to liver disease, Alagille syndrome is associated with cardiac disease, skeletal and ocular anomalies, and characteristic facies. Molecular testing has shown that the frequency of the pathogenic JAG1 mutation is 1 in 30,000 [24]. While many patients present with cholestasis before the age of one, patients may present at an older age as well. Cholestatic pruritus and xanthomas are associated with chronic cholestasis in Alagille syndrome. Laboratory findings are often remarkable for serum bilirubin values up to 30 times the normal and serum bile salts up to 100 times the normal [1].
DIAGNOSTIC APPROACH
As previously discussed, prompt evaluation should be initiated for a child with suspected cholestasis as exhibited by clinical appearance of jaundice, dark urine, and acholic (unpigmented) stools. The right upper quadrant should be evaluated for liver size and texture. Hepatosplenomegaly may be a clinical finding on examination, depending on the etiology of the cholestasis. A firm, nodular liver can be a clue to the development of cirrhosis and portal hypertension from long-standing cholestasis. In addition to liver abnormalities, other stigmata indicative of cholestasis and secondary chronic liver disease include ascites, palmar erythema, caput medusae, spider angioma, gynecomastia, and splenomegaly.
Initial evaluation in children with jaundice can be made with a urine dipstick for bilirubin and serum total unconjugated and conjugated bilirubin levels [1]. The presence of bilirubin in the urine confirms conjugated hyperbilirubinemia, since unconjugated bilirubin is not excreted in the urine. This can be very helpful as bilirubinuria can be detected before clinically observable jaundice [1].
There are several laboratory methods used to determine conjugated bilirubin level. The preferred method secondary to more accurate and sensitive quantification of bilirubin is via chromatographic analysis [25]. When analyzing serum bilirubin, it is vital to keep note of two details: magnitude of bilirubin increase and presence of delta-bilirubin. The magnitude of increase of serum bilirubin does not differentiate between intrahepatic and extrahepatic biliary diseases, as both conjugated and unconjugated bilirubin are retained and a wide margin of serum concentrations can be observed [1]. Delta-bilirubin is the fraction of bilirubin irreversibly covalently bonded to albumin in serum. It is metabolized with albumin and thus has a half-life of 14 days [26]. Delta-bilirubin often prolongs direct hyperbilirubinemia, while other tests may be normalizing, as it is larger in size, protein-bound, and not easily excreted in bile or urine [12627]. Some laboratories estimate the direct bilirubin, which includes delta-bilirubin, and can overestimate the true level of conjugated hyperbilirubinemia.
The work up for cholestasis is broad due to the large differential (Fig. 1). Evaluation of the etiology of cholestasis can be divided into initial testing and additional laboratory and radiology evaluation (Table 1).
In the primary investigation of cholestasis, it is important to rule out infectious etiologies. The association between sepsis and cholestasis has well elucidated pathophysiologic mechanisms. The liver responds to infection by increasing production of vital amino acids, lipids, complement, C-reactive protein, and other secretory substances. The same cytokines that activate mobilization of liver resources during the acute phase response play a role in suppression of expression and function of hepatobiliary transporters.
Bacterial infections can cause conjugated hyperbilirubinemia through the effect of circulating lipopolysaccharide (LPS) endotoxin within the liver. At a hepatocellular level, LPS endotoxin binds to membrane receptors on Kupffer cells and stimulates them to release pro-inflammatory cytokines, leading to downregulation of bile acid transporters [8]. Urinalysis and urine culture, blood culture, and viral studies are initial laboratory investigations that should be obtained to rule out serious infections in the setting of acute onset cholestasis.
A more common infectious cause of cholestasis is viral hepatitis. Diagnosis of viral hepatitis relies on serologic markers for hepatitis A. Chronic hepatitis B, and C viruses rarely cause cholestasis until later stages of disease. A myriad of other viruses, such as Ebstein-Barr, cytomegalovirus, adenovirus, influenza virus, parvovirus, enteroviruses, herpes simplex, and varicella virus can also cause cholestasis due to hepatic involvement [28].
Initial evaluation should also include enzymatic measures of cholestasis as well as liver function tests. These include GGT level, alkaline phosphatase, serum aspartate aminotransferase, and alanine aminotransferase levels. The liver is the source of protein synthesis. Chronic cholestasis is associated with decreased production of clotting factors and other proteins. Laboratory findings will demonstrate low albumin and elevated international normalized ratio and prolonged prothrombin time.
Chronic liver disease reduces the function and synthesis of insulin-like growth factor 1 (IGF-1), IGF binding protein 3, and growth hormone (GH) levels. Typically IGF binding proteins 1 and 2 are increased. GH resistance present in chronic liver disease and children do not respond to GH administration [2930]. As a result, children with chronic liver disease may develop short stature and are at risk for decreased brain growth, impaired mental development, and immunosuppression [1]. If chronic cholestasis occurs, these children are also at risk for fat-soluble vitamin deficiency. Therefore, evaluation and monitoring should be done for fat-soluble vitamins A, D, E and K.
Anatomical evaluation should be performed early in the diagnostic workup. Because the most common causes of biliary obstruction in the pediatric age group are cholelithiasis and choledocholithiasis, ultrasound is the first line recommended imaging modality. Ultrasound is highly sensitive and specific for gallstones >1.5 mm in diameter, with decreasing test sensitivity with stones located in the common bile duct [3132]. ERCP is the gold standard for the investigation and treatment of common bile duct stones (Fig. 3).
Trans-abdominal ultrasonography can reveal biliary cysts, and has a sensitivity of 71-97% [33]. This imaging modality is limited by body habitus, bowel gas, and overlying structures. If a cyst is suspected on ultrasound, cross-sectional imaging is the next step. Although CT cholangiography may be utilized, magnetic resonance cholangiopancreatography (MRCP) is the gold standard [34]. CT cholangiography has a high sensitivity of around 93% for visualizing the biliary tree, 90% for biliary cysts, and 93% for intraductal stones. However, it is only 64% sensitive for viewing the pancreatic duct [32]. MRCP is a favorable imaging modality as it uses non-ionizing radiation and allows for the visualization of obstructive lesions within the biliary tree as well as the pancreas. It is 73-100% sensitive for detecting biliary cysts [35]. If concerns remain for obstruction following MRCP, ERCP may be used as a diagnostic and therapeutic tool. ERCP or MRCP is used to image the bile ducts and shows strictures and dilations of affected portions of the biliary system [31].
Second line evaluation of cholestasis should be targeted to uncommon diseases of metabolism, mitochondria, and genetics. An ammonia level, serum amino acids, acyl carnitine profile, urine organic acids, lactate, and pyruvate may be indicated. Auto-antibody testing should be considered if the patient has a positive family history for autoimmune disease.
Serologic autoantibody tests can be used for diagnosis, and include immunoglobulin G levels, anti-nuclear antibody, and anti-smooth muscle antibody. However, for autoimmune hepatitis, a liver biopsy is the gold standard. This typically shows piecemeal necrosis of hepatocytes, dense inflammatory infiltrate in the liver, and surrounding mononuclear and plasma cells.
Primary sclerosing cholangitis (PSC), an autoimmune disease, is diagnosed based on biochemical, histologic, and radiologic findings. Destruction of intra and extrahepatic bile ducts is characteristic of PSC and is visualized on MRCP and ERCP. Both modalities can reveal a beading appearance consistent with regions of luminal narrowing and bile duct dilation. Sensitivities are 90% for MRCP and have been reported to be even higher for ERCP [3637]. However, MRCP is better at visualizing bile ducts proximal to obstructed regions. Laboratory findings such as elevated alkaline phosphatase or GGT are common. Conjugated bilirubin is elevated in severe disease [38]. Histological findings are not specific, and biopsy is not always diagnostic. The classic "onion skinning" appearance of edema, and fibrosis around interlobular bile ducts are pathognomonic for PSC. However, nonspecific features such as the presence of mononuclear cells and ductular proliferation may also be observed [1].
Additional evaluation for rarer etiologies of cholestasis such as PFIC may be indicated. As previously discussed, PFIC types I and II are associated with normal GGT levels and elevated serum bile salts, whereas type III presents with elevated GGT level. DNA sequencing and genetic testing for mutations in the ATP8B1, ABCB11, and ABCB4 genes can confirm a diagnosis of PFIC types I, II, and III respectively [39].
A diagnosis of Wilson's disease can be made with measurement of low serum ceruloplasmin, 24 hour urine copper, ophthalmologic examination for Kayser- Fleischer rings (late finding), and liver biopsy for elevated hepatic copper concentration ≥250 µg/g dry weight [40]. In addition genetic testing exists for the diagnosis of Wilson's disease.
MANAGEMENT
The initial therapy for cholestasis is treating the underlying etiology of the disease. Ursodeoxycholic acid, a potent choleretic, has been shown to alleviate pruritus and improve liver function tests, including bilirubin, alanine aminotransferase, and cholesterol in multiple randomized, placebo controlled trials [3].
In addition, some chronic cholestatic liver diseases (such as Alagille syndrome) are linked with the findings of nutritional deficiencies and pruritus. The authors' goal is to review updated treatment of pruritus and nutritional deficiencies.
Treatment of pruritus
Pruritus is a common challenging side effect of many hepatobiliary syndromes. It generally affects the distal extremities, including the palms and soles. Pruritus is reported by patients to have a diurnal variation of itch severity, with worsening of symptoms in the late evening [42]. It can negatively affect a patient's appetite, sleep, and cognitive development and thus adversely affect his or her quality of life. While the pathogenesis of pruritus associated with liver disease is not well elucidated, new management strategies have been recently described in the literature to ameliorate the burdens of pruritus [43].
Current first line management is aimed at removing the pruritogen by decreasing the bile acid concentration through medical or surgical management. First line therapy is cholestyramine, a bile acid resin that blocks the absorption of bile in the terminal ileum [44]. As monotherapy has often been reported to be ineffective, combination therapy is often beneficial (Fig. 4) [4]. Second line agents include rifampicin, a hepatic enzyme inducer, which enhances the metabolism of pruritogenic substances and induces hydroxylation and excretion of bile acids via the kidneys [442]. Third line treatment is aimed at antagonizing opioid receptors which are upregulated in cholestasis with the use of agents such as naloxone or naltrexone [44]. In the older adolescent and young adult, a fourth line agent, Sertraline, a selective serotonin uptake inhibitor, can also be considered because it has been shown to have moderate antipruritic effects [45]. Of note, ondansetron, a 5-HT3 antagonist, has also been shown to have significant positive effect as compared to a placebo [46]. Surgical management is invasive and includes nasobiliary and transcutaneous drainage or external biliary diversion [2]. Surgical management is reserved for refractory and desperate cases.
Treatment of nutritional deficiencies
1. Energy
Children with chronic liver disease have elevated resting energy expenditure, reported to be 30% greater than normal [47]. This is why children with hepatic disease benefit from caloric supplementation.
2. Proteins and branch chain amino acids (BCCA)
Disordered carbohydrate metabolism results in amino acids being used for gluconeogenesis. There is also increased protein catabolism in patients with cholestasis. A protein intake of 4 g/kg of body weight is necessary and does not precipitate encephalopathy. In addition, there is an increased requirement of branched chain amino acids (leucine, isoleucine, and valine), which are essential amino acids. In normal healthy children, the requirement is 147 mg/kg per day, as opposed to a patient with hepatic disease whose demand is 207 mg/kg per day [48]. It is therefore important to supplement the diet with proteins, especially BCCA.
3. Fats and fat soluble vitamins
In cholestasis there is impaired digestion and absorption of long chain triglycerides [47]. Medium chain triglycerides are absorbed in the absence of micelles. Since vitamins A, D, E and K require micelles, they also become deficient in cholestatic diseases [47]. Fat-soluble vitamin insufficiency is seen when total bilirubin is greater than 2 mg/dL [4849]. Several studies have shown that in cholestatic illnesses, it is difficult to maintain adequate serum fat-soluble vitamin levels [495051]. In a recent study published in 2012 by Shen et al. [50], patients with cholestatic disease receiving conventional multivitamin supplementation still demonstrated significant fat-soluble vitamin deficiencies. Work published by Jensen et al. [51] in 2015, illustrates that even doses of 150,000 IU daily of ergocalciferol (vitamin D2) taken for 2-3 days and 4 week follow up failed to achieve sufficient levels (>20 ng/dL) in cholestatic patients.
As a result, these vitamin levels should be monitored and supplemented separately [50]. Often, vitamin A is cautiously supplemented secondary to an adverse effect including hepatotoxicity. Of note, co-administration of micellar vitamin E or aqueous vitamin E formulations may improve vitamin D absorption in children with cholestasis [47]. Absorption of oral vitamin E can be improved by using the form d-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) [47]. For vitamin K, oral and parenteral dosages can be used, although absorption of enteral supplements can be poor [47]. In addition, essential fatty acids (linoleic and linolenic acids found in vegetable oils) should be monitored and supplemented [4750].
4. Water soluble vitamins and trace elements
In addition, the literature recommends supplementation of water-soluble vitamins and trace elements in patients with chronic liver disease. This includes B complex vitamins, vitamin C, selenium, and zinc [47]. Patients with cholestasis have also been shown to have elevated serum levels of copper and manganese, because these metals are typically excreted in bile [47]. Caution should be used in patients with cholestasis receiving total parenteral nutrition with additives of copper and manganese [47].
CONCLUSION
While cholestasis has numerous etiologies, understanding the pathophysiology of cholestasis and the complexity of bilirubin metabolism is integral to understanding the etiology of the disorder. Given the broad differential, a stepwise evaluation approach is essential. Once a diagnosis is established, treatment is tailored based on the etiology of the disease. In cases where the cholestasis is caused by an infectious etiology, proper medical management is initiated. However, cholestasis can often be chronic and management may need to focus on alleviating negative side effects. Optimal medical management of pruritus and nutritional deficiencies is imperative when the underlying etiology cannot be reversed.


 XML Download
XML Download