

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Inflammatory myofibroblastic tumor (IMT) is rare mesenchymal solid tumor that consists of proliferating myofibroblasts with an inflammatory infiltrate background. It occurs mainly in children and young adults, but has a low prevalence in infants [1,2]. IMTs may be located in various organs, mainly those in the thorax, but intra-abdominal lesions are relatively rare [3].
Intra-abdominal IMTs have clinical importance due to their relationship to malignancy. Intra-abdominal IMTs are difficult to distinguish preoperatively from other malignancies, such as sarcomas, lymphomas, and metastases [4]. IMTs were originally reported to be benign tumors, but can exhibit locally aggressive neoplastic processes and metastases similar to malignancies. Based on such findings, surgical excision of IMTs is recommended [2].
In this report we describe two cases of infantile intra-abdominal IMT that were histopathologically diagnosed following surgical excision.
CASE REPORT
Case 1
A 4-month-old male infant was transferred to our institution for further evaluation of a palpable upper abdominal mass. The mass was found incidentally during the screening for a regular vaccination. He was vaginally delivered at 37 weeks of gestation and weighed 3,260 g. He had no previous medical or familial history. Physical examination revealed a soft, non-tender mass in the right upper quadrant of the abdomen.
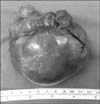
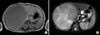
Computed tomography scan revealed a 7.5×6.8×6.3 cm cyst-like mass attached to the small intestine and located in the right abdomen, inferior to the liver and anterior to the kidney (Fig. 1). We performed the laparoscopic exploration to evaluate the mesenteric cyst. An 8.5×7.2×6.3 cm solid, not cystic, mass was located at the ileal mesentery. Tumor excision with preservation of the ileum was performed (Fig. 2).
Histological examination revealed a proliferation of spindled myofibroblasts in a background with infiltration of chronic inflammatory cells such as lymphocytes and plasma cells. On immunohistochemical staining, the tumor cells showed cytoplasmic positivity for desmin, smooth muscle actin (SMA), D2-40, and anaplastic lymphoma tyrosine kinase receptor (ALK) 1 and negativity for HMB45, c-kit, S-100, Factor VIII, CD34, CK5/6, h-caldesmon, epithelial antigen membrane, pan-CK, and calretinin (Fig. 3).
His postoperative course was uneventful. The patient is currently doing well with no evidence of recurrence during the 3 months of follow-up.
Case 2
A 5-month-old male infant was referred to our hospital for further evaluation of a hepatomegaly found during the screening for a regular vaccination. He was born at 40 weeks of gestation via Cesarean section and weighed 3,380 g. He had no previous medical or familial history. Physical examination revealed a hepatomegaly for which the liver margin was palpable by 6 cm below the costal margin without pain or tenderness.
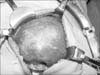
Ultrasound scan showed about 10 cm-sized mass in right lobe of liver. For evaluating the relationships with neighboring structures and differential diagnosis, magnetic resonance image (MRI) was conducted. MRI showed a 10.4×9.5×9.0 cm mass in the right lobe of the liver. The mass exhibited low signal intensity on T1-weighted images, heterogeneous signal intensity on T2-weighted images, and positive enhancement and diffusion restriction on diffusion-weighted images (Fig. 4). We assumed the mass was a benign hepatic mass such as focal nodular hyperplasia or mesenchymal hamartoma. Sono-guided biopsy was performed and IMT was diagnosed histologically. At operation, a solid mass measuring 11.5×9.5 cm was presented at the right lobe of the liver (Fig. 5) and right hemihepatectomy was performed.
Pathological examination revealed spindle cell proliferation with infiltration of lymphocytes and plasma cells, compatible with IMT. On immunohistochemical staining, the tumor cells showed positivity for ALK and SMA, focal weak positivity for c-kit, and negativity for desmin, h-caldesmon, S-100, CD21, and CD34.
His postoperative course was uneventful. The patient is currently doing well with no evidence of recurrence during the 14 months of follow-up.
DISCUSSION
IMT has been described with a variety of terms in the past [5]. However, it is generally recognized as a neoplasm with the possible presence of local recurrence, multifocality, and distant metastasis. Because IMT has distinctive histopathologic features, comprising myofibroblastic mesenchymal cells mixed with an inflammatory infiltration of lymphocytes, plasma cells, and histiocytes, it has been designated as a specific entity. Although it usually occurs in the lung, the extra-pulmonary region, such as head and neck, abdominopelvic cavity, retroperitoneum, and rarely solid organs in the abdomen, can be involved. Consequently, IMT can present with variable clinical symptoms according to the site of tumor origin. IMT is primarily a soft tissue tumor in young adults, most frequently occurring in the first two decades of life, but IMT may occur over a wider age range (3 weeks to 74 years) [2,5,6,7]. In Korea, there have been few reported infantile cases with the youngest patient being a 3 month-old infant with IMT at the transverse colon mesentery [8].
The etiology of IMT has not been fully described and is currently regarded as a specific neoplastic lesion with inflammatory sclerosing and fibrosing processes that may be recognized by cytogenetic aberrations such as ALK gene rearrangement, p53 mutation, and MDM2 expression [3,7]. Those associations may act as prognostic indicators that could suggest the need for aggressive clinical behavior, particularly when the IMT exhibits a combination of cellular atypia, ganglion-like cells, aneuploidy, and p53 overexpression [3,9].
Although the appropriate treatment for IMT is variable, surgical excision has been the cornerstone in managing this tumor. In one case, when complete resection of a hepatic IMT was impossible, total hepatectomy and liver transplantation were performed [3]. Other authors have suggested the use of steroids, radiotherapy, chemotherapy, and non-steroidal anti-inflammatory drugs, especially COX-2 inhibitors as an adjuvant therapy, but there is no consensus on an effective non-surgical therapy [2,10].
ALK expression is found in 36-60% of IMT and considered that having relations with aggressive behaviors, including more frequent intraabdominal location and epithelioid tumoral morphology, larger tumor size, and higher recurrent rate. But, though ALK expression is positive, complete surgical resection is known to be able to improve the prognosis [11,12].
In our cases, two different abdominal IMT types were described: one originating in mesentery and the other in a solid organ (liver). Fortunately, complete resection was successful in both cases done. In IMT, recurrence and metastasis are rare and they have been associated with certain conditions, including more aggressive behavior and multifocal tumors, retroperitoneal location, infiltration of adjacent structures, and incomplete excision [5]. In our cases there has been no recurrence.
In summary, examples of abdominal IMT are infrequent in infants and our two cases were successfully treated by complete surgical resection. Considering the biological behavior of the intermediate type of neoplasm in IMT, we expect good survivals when achieving appropriate surgical resection without adjuvant therapy.


 XML Download
XML Download