

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Congenital chloride diarrhea (CLD) is a rare inherited autosomal recessive disorder [1]. Solute carrier family 26 member 3 (SLC26A3) gene encoding chloride ion (Cl-)/bicarbonate (HCO3-) exchanger is responsible for the acidic, Cl- rich diarrhea [2]. Chronic profuse diarrhea causes dehydration and the renin-angiotensin system is also activated. This results in hypochloremia, hyponatremia and metabolic alkalosis with dehydration. Severe dehydration and electrolyte imbalance may lead to lethargy and death if untreated, but early appropriate supplementation therapy can help normal growth and favorable outcome [3].
CASE REPORT
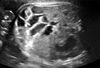
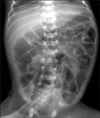
A 6-month-old male presented to our outpatient clinic with poor feeding and irritability. He was the second of non-identical twins, born by cesarean section at 32 and 2/7 weeks gestational age due to preterm labor and transverse lie of one fetus. Since their mother had polyhydramnios and both showed small bowel dilatation on prenatal ultrasound (Fig. 1), both newborns were suspected to have congenital intestinal obstruction and their mother was transferred to this hospital before delivery. Amnioreduction has been conducted twice at previous hospital but there were no data for diagnosis such as amniotic fluid analysis. The birth weight of the male twin was 2,130 g and that of the female was 1,720 g. They were treated in the neonatal intensive care unit (NICU) for prematurity and low birth weight. During that time, they showed recurrent abdominal distension and ileus, but there was no evidence of bowel obstruction on either abdominal sonography or small bowel series (Fig. 2). They were discharged at 42 day of age.
One month later, the male was admitted for poor weight gain. Sucking was insufficient and he was irritable. He had hyponatremia and metabolic alkalosis. Blood gas analysis showed a pH of 7.645 and a base excess of +16.0 mmol/L. Other laboratory findings were serum sodium 115 mEq/L, serum potassium 2.6 mEq/L, serum chloride 57 mEq/L. urine sodium 8 mEq/L, urine potassium 3.2 mEq/L, urine chloride 9 mEq/L, urine osmolality 61 mOsm/kg H2O, and serum osmolality 235 mOsm/kg H2O. Renin activity (116.89 ng/mL/h; normal range, 2.0-35.0 ng/mL/h) and aldosterone level (>1,393 pg/mL; normal range, 65-860 pg/mL) were elevated. He had watery diarrhea, but stool culture and stool occult blood examinations were negative. Electrolytes were corrected and symptoms improved. He was discharged but the cause of hyponatremia and metabolic alkalosis was not clear.
The infant was admitted at 6 months of age because of diminishing body weight and developmental delay. He was only able to consume milk and could not eat solid food. He was unable to lift his head or roll over, did not grasp anything and rarely laughed or babbled. His body weight was 3.6 kg (corrected age, 4 months; below the 3rd percentile) and his height was 60 cm (corrected age, 4 months; 5th to 10th percentile). He again had hyponatremia and metabolic alkalosis. Urine osmolality was elevated and urine sodium was low: urine sodium <10 mEq/L, urine potassium 8.9 mEq/L, urine chloride 8 mEq/L, urine osmolality 390 mOsm/kg H2O and serum osmolality 215 mOsm/kg H2O. Hypotonic hyponatremia with urine osmolality >100 mOsm/kg, decreased extracellular fluid volume, and urine sodium <10 mEq/L suggest presence of extrarenal solute loss. Metabolic alkalosis with low urine chloride (<10 mEq/L) suggest vomiting, gastric drainage, diuretic use, or chloride-losing diarrhea, and other common misdiagnosis such as Bartter's syndrome could be excluded. This results prompted evaluation of fecal electrolytes. He had been experiencing numerous daily episodes of watery stool. Stool Cl- and pH was 124 mEq/L and 6.18, respectively. He was diagnosed as CLD (Table 1).
The patient had been born to non-consanguineous parents. His twin sister was doing well after discharge from NICU but she also had frequent diarrhea and growth retardation (corrected age, 4 months; body weight 5.1 kg, below the 3 percentile) and her twin brother was diagnosed with CLD. She was also underwent some blood tests with exams on stool electrolyte, confirming her with CLD as well. She showed relatively minor abnormality in electrolyte level of serum and stool than her twin brother (Table 1).
At the present time, 13 months after diagnosis, both twins are orally receiving NaCl and KCl. The dose of NaCl and KCl dosage were adjusted with constant monitoring of the patients' electrolyte level. In the last outpatient clinic, the dosage was NaCl 4.2 mEq/kg/day, KCl 0.8 mEq/kg/day for the male, and NaCl 3.2 mEq/kg/day, KCl 1.2 mEq/kg/day for the female patient. The male's body weight is 10.5 kg (corrected age, 17 mo; 25th to 50th percentile), serum sodium is 141 mEq/L, serum potassium is 4.3 mEq/L and serum chloride is 105 mEq/L. The female's body weight is 10.2 kg (corrected age, 17 months; 25th to 50th percentile), serum sodium is 141 mEq/L, serum potassium is 3.7 mEq/L and serum chloride is 103 mEq/L. Their serum electrolytes are being maintained within normal range and their body weights and developments are gradually improving (Fig. 3). Gene study was recommended, but parental consent was not given.
DISCUSSION
CLD is a rare diarrheal disease inherited in an autosomal recessive manner [1]. The gene for CLD, SLC26A3, resides on chromosome 7q31 [4]. Over 30 mutations of CLD have been identified, there are several founder mutations and many different minor mutations. The majority of Finnish patients have V317del, Arabic patients tend to have G187X and many Polish patients have I675-676ins mutation [4]. The various mutations are not manifested as different phenotypes.
Diagnosis of CLD is based on its clinical picture and fecal electrolytes. The typical features can enable a prenatal diagnosis [10]. Fetal bowel loops are dilated and fetal diarrhea causes polyhydramnios. This prenatal finding is often considered as evidence of fetal intestinal obstruction.
Profuse diarrhea of the infant leads to dehydration, weight loss, metabolic alkalosis, electrolyte imbalance and sometimes death. If proper treatment is delayed, chronic dehydration and electrolyte imbalance may result in developmental retardation, chronic kidney disease and male subfertility [11]. Early diagnosis and early treatment is essential. As CLD is rare and more than half of patients are found in Finland, Poland and Arab countries, delayed or missed diagnosis is possible in other countries [12].
Classical treatment is oral or intravenous replacement of NaCl and KCl [11]. New methods being developed include proton pump inhibitors [13], butyrate [14] and cholestyramines [11]. Long-term prognosis is favorable if diagnosed early and managed properly. Continuous consumption of salt substitutes and regular follow-up throughout the life are needed.
In 1945, Gamble et al. [15] and Darrow [16] first described congenital alkalosis with diarrhea. In 1988, Lee et al. [17] reported the first two cases in Korea. Several patients have been were reported in Korea; all were misdiagnosed as chronic diarrhea, Bartter syndrome, Reye syndrome, Hirschsprung disease or small bowel obstruction. One premature infant presumed as small bowel obstruction underwent an exploratory laparotomy [18]. In Korea, no CLD patient has undergone genetic study, but the first mutational analysis was conducted in 2012. A missense variant (c.525G>C; p.Arg175 Ser) and a splicing mutation (c.2063-1G>T) in the SLC26A3 gene were identified in that patient [19]. Recently, genetic analysis was performed in eight Korean-origin patients with non-consanguineous parents, and the most common mutation found was c.2063-1G>T [20].
To the best of our knowledge, this is the first case report of non-identical twins affected by CLD. Despite being twins, the severity of symptoms differed. There are several cases of sibling with CLD but there are little data about the different clinical courses of twin patients. Despite same type of genetic mutation, many patients with CLD present with varying severity. Patients with CLD are vulnerable to gastrointestinal tract infection. Whether these twins have the same genetic defects or not, in our opinion, infection or other factors may have made such difference in their clinical courses and outcomes. The male had experienced severe metabolic alkalosis and hyponatremia that led to developmental delay and growth retardation. Early diagnosis and proper treatment could prevent these crisis and complications. We cannot overemphasize the importance of early recognition and early treatment of CLD. We could not ascertain the causative mutations since parental consent for a gene study was not given. If mutations are identified, it would be helpful to understand the incidence of disease and counseling. But even before carrying out genetic study, we should be aware of the possibility of CLD when encountering a patient with chronic diarrhea. If neonates show prenatal findings of dilated bowel loops and polyhydramnios, but there is no evidence of intestinal obstruction after birth, we should be mindful of CLD and consider evaluating stool electrolytes.


 XML Download
XML Download