

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Down syndrome is the most common chromosomal abnormality recognized at birth, and is associated with many anomalies of organ systems including the cardiac, hematologic and gastrointestinal systems. Down syndrome is a rare cause of neonatal cholestasis and the association with neonatal cholestasis has not been well-substantiated. There have been several reported cases of neonatal cholestasis in Down syndrome associated with bile duct paucity, myeloproliferative disorders and neonatal hemochromatosis. The liver diseases in these cases were fatal [1]. To the best of our knowledge, until now benign neonatal cholestasis associated with Down syndrome has not been reported.
We report our experience with a case of idiopathic neonatal cholestasis that spontaneously resolved in a patient with Down syndrome.
CASE REPORT
Patient: A one-day-old female
Chief complaint: Hepatosplenomegaly on prenatal ultrasonography
Present illness: The patient was born at 386/7 weeks of gestation via normal vaginal delivery. The patient was admitted to the neonatal intensive care unit in our hospital because of hepatosplenomegaly on prenatal ultrasonography.
Past medical and family history: The patient's prenatal examination did not show any specific abnormal findings except hepatosplenomegaly on a prenatal ultrasonography conducted 1 month prior to delivery. The patient's mother was a healthy, 26-year-old woman without any medical problems or use of any medications except iron supplementation. The parents of the patient were not consanguinities and a 3-year-old female sibling was healthy.
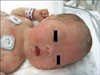
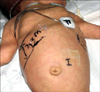
Physical examination: The patient's Apgar scores were 6 at 1 minute and 9 at 5 minutes. At the time of birth, the pulse rate was 116 beats/minute, respiratory rate was 42 beats/minute, body temperature was 36℃ and oxygen saturation was 75% on oximetry at room air. Oxygen saturation recovered to 93% at room air at 10 minutes after birth. The birth weight was 2,370 g (<10 percentile), height was 49.5 cm (50 percentile) and head circumference was 32 cm (<10 percentile). Physical examination revealed a flat face, low nasal bridge, protruding tongue and short neck (Fig. 1). Simian creases were evident on both palms. Hepatosplenomegaly was detected on abdominal palpation. We could palpate the liver about 3 cm below the costal margin and also palpate the spleen about 2 cm below the left costal margin on abdominal examination (Fig. 2). There was no abdominal tenderness. A hypotonic posture was evident.
Laboratory and radiologic findings: Initial complete cell counts were as follows: hemoglobin 18.9 g/dL; hematocrit 58; white blood cell count 10,170/mm3 and a platelet count 105×103/mm3. C-reactive protein was 0.2 mg/dL (normal range, 0.0-5.0 mg/dL). Coagulation tests were within normal limits for this age. The platelet counts fluctuated between 53×103/mm3 and 267×103/mm3. Initial laboratory findings were as follows: aspartate aminotransferase (AST) 57 U/L (normal ranges at this age, 35-140 U/L); alanine aminotransferase (ALT) 50 U/L (normal range, 6-50 U/L); total and direct bilirubin 4.16 and 1.51 mg/dL, respectively; ammonia 61 µmol/L (normal range, 21-95 µmol/L); creatine kinase 244 U/L (normal range, 214-1,175 U/L) and lactate dehydrogenase 594 U/L (normal range, 170-580 U/L). Gamma glutamyl transpeptidase (GGT) was 274 U/L (normal range, 13-147 U/L) at 5-days-of-age and α-fetoprotein level was 43,896 ng/mL. The level of α-1 antitrypsin and bile acid in plasma were within normal limits (194 and 89.3 µmol/L, respectively). Serologic tests as IgM for toxoplasma, rubella, cytomegalovirus, herpes virus, varicella zoster virus, hepatitis C virus, hepatitis A virus and hepatitis B surface antigen were all negative. Thyroid function tests were all within normal limits. Expanded neonatal screening test by tandem mass spectrometry was done at 20 day of admission and the results were within normal limits. Cultures of body fluids including blood, urine, stool and cerebrospinal fluid were all negative during hospitalization. Trisomy 21 was proven on chromosomal study and direct sequencing of the GATA1 gene was normal.
Abdominal ultrasonography was performed at day 2 following admission due to hepatosplenomegaly, which also revealed hepatosplenomegaly and a prominent hepatic artery.
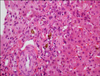
Pathologic findings: We performed biopsies of liver and bone marrow on days 29 and 33 of admission, respectively. On pathologic examination of liver, minimal lobular activity and severe cholestasis were noted (Fig. 3). But, there were no fibrosis, cellular infiltration or abnormal biliary tract on light and electron microscopy examination. On bone marrow examination, three or four megakaryoblasts were observed in one high power field. However the finding was not sufficient for a diagnosis of myeloproliferative disorder.
Hospital course: At the time of admission, liver function tests were within normal limits. But, serum total and direct bilirubin rose to 13.81 mg/dL and 2.01 mg/dL, respectively, without hemolysis on day 2 of hospitalization. Hemoglobin was 18.9 g/dL at the same time. Serum direct bilirubin rose slowly up to 5.42 mg/dL by day 20 of hospitalization (Fig. 4), but the stool was yellowish during this period.
The level of GGT increased from 274 U/L at 5-days-of-age to 438 U/L at 1-month-of-age. The levels of AST and ALT fluctuated (Fig. 4). A second abdominal ultrasonography was performed at day 20 of hospitalization, which revealed hepatomegaly without structural abnormality of biliary tract. The splenomegaly and prominent heptic artery that was evident on the first ultrasonography was not found. We performed liver and bone marrow biopsy after about 1-month of hospitalization because of sustained direct hyperbilirubinemia and thrombocytopenia.
The decision based on the above laboratory, radiologic and pathologic findings was for conservative care with carefully observation of clinical manifestations. The patient received urosdeoxycholic acid (10 mg/kg/day), fat soluble vitamins and was fed protein hydrolysate formula. By 2-months-of-age, the direct hyperbilirubinemia, thrombocytopenia and hepatosplenomegaly were improved. The levels of total bilirubin and platelet counts were 0.49 mg/dL and 225×103/mm3, respectively.
DISCUSSION
There are many causes of cholestasis in young infants. Early diagnosis and initiation of appropriate therapy is important, because the effects of this disorder are usually very serious. Down syndrome is also one of the causes of infantile cholestasis, but is very rare. Cholestasis associated with Down syndrome is not well understood and had a fatal clinical course in reported cases [1,2]. The liver diseases in those cases were associated with myeloproliferative disorder, hemochromatosis and bile duct paucity. Several mechanisms in liver disease associated with Down syndrome that have been suggested are a higher concentration of superoxide dismutase (SOD), higher incidence of viral hepatitis and extramedullary hematopoiesis [1].
In our case, there was no evidence of these mechanisms. An elevated SOD concentration causes continuous radical injuries and iron overload. In other words, increased amount of SOD could be followed by an imbalance in scavenging oxidative stress including glutathione peroxidase and catalase. Imbalance in scavenging oxidative stress may have deleterious effects on oxidative processes including lipid peroxidation. Iron overload may be associated with an increased lipid peroxide formation [3,4]. Although we did not check the patient's iron panel during the hospitalization period, we could rule out neonatal hemochromatosis through the patient's clinical course, coagulation test, α-fetoprotein and liver pathology. The patient also had no indications of any congenital viral hepatitis or extramedullary hematopoiesis on clinical, laboratory and pathologic examinations.
Chronic or acute perinatal distress also may transiently cause neonatal cholestasis, regardless of Down syndrome. Clinically, transient neonatal cholestasis is characterized by early onset of cholestasis, lack of known causes of neonatal cholestasis after careful workup is carried out, spontaneous normalization of clinical and biochemical parameters of liver function during follow-up and presence of several predisposing events possibly responsible for perinatal distress. Perinatal insults can lead to hepatic hypoxia or ischemia and can disturb bile secretion [5].
In our case, the patient displayed relatively good Apgar scores. But, the patient was small for gestational age. Chronic or acute ischemia or hypoxia of the liver in utero as a result of intrauterine growth retardation is a possibility. In addition, bile secretion processes may also be impaired by hepatic hypoxia-ischemia [6]. In a rat model of hepatic hypoxia-ischemia, cholestasis may be caused by biliary canalicular injury or bile duct alteration, which may be paradoxically exacerbated after reperfusion of blood or oxygen to previously ischemic or hypoxic liver [7]. Pathologic examination of the patient's liver biopsy specimen revealed disturbed bile secretion without biliary tract abnormalities.
There was a prominent hepatic artery evident on the first abdominal ultrasonography, which might not affect significantly hepatic hypoxia or ischemia, since the hepatic blood supply generally depends on the portal vein. Two abdominal ultrasonographies did not show a triangular cord sign. But, a periportal echogenecity might not be shown on an ultrasonography in early stage of biliary atresia and, even though there are no triangular cord signs, biliary atresia cannot be ruled out [8]. We did not perform hepatobiliary scintigraphy because of its low specificity, high false positive rate [9] and the patient's yellow stool.
We performed liver biopsy to rule out extrahepatic biliary atresia, biliary paucity, hemochromatosis and myeloproliferative cell depositions because we could not find clear causes of the cholestasis on careful workups. We could rule out the aforementioned severe diseases based on the patient's clinical course and examinations, and initiated conservative care with careful monitoring. Cholestasis improved slowly and the patient was discharged at 2-months-of-age.
We tentatively suggest that another unknown mechanism in Down syndrome and chronic hepatic hypoxia or ischemia in utero under the detected level might affect cholestasis in our case. Further study about cholestasis in Down syndrome may be warranted.
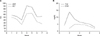
 XML Download
XML Download