

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Peutz-Jeghers syndrome (PJS) is a rare autosomal dominant genetic disease that is characterized by multiple gastrointestinal hamartomatous polyps and mucocutaneous melanocytic macules [1]. Patients with PJS have an increased risk of developing various benign and malignant neoplasms of the gastrointestinal and extra-intestinal organs [1]. Especially, women with PJS usually have an increased tendency for developing breast and gynecologic neoplasms [2,3]. In particular, minimal deviation adenocarcinoma (MDA) of the cervix, and sex cord stromal tumors with annular tubules (SCTATs) of the ovaries are rare but commonly associated with PJS [2-4].
In 1998, a serine threonine kinase (STK11) gene, also known as LKB1, at chromosome 19p 13.3 was reported to be defective in PJS [5]. The STK11/LKB1 gene encodes for a member of the serine/threonine kinase family, regulates cell polarity, and functions as a tumor suppressor gene. Mutations of STK11/LKB1 in malignant tumors accompanied by PJS have relevance to the aggressive clinical course reported in some studies [3].
We herein report two cases of MDA of the cervix and tumorlets of SCTATs of the ovary associated with PJS. A frame shift mutation of codon 132 in exon 7 of STK11/LKB1 was found in case 1.
The patient with the mutation of STK11/LKB1 showed a fatal clinical course, and although additional combined chemo-radiation therapy was applied, she expired 1 year later. However, another patient without the mutation has shown no clinical evidence of recurrence to date.
CASE REPORTS
1. Case 1
A 36-year-old single, virgin woman presented with lower abdominal pain and hypermenorrhea for 2 months. Her past medical history was unremarkable. There were several melanin pigments on the tips of the patient's fingers and lips. The gynecological examination was not performed. Pelvic magnetic resonance imaging (MRI) revealed a 5.6×4.8×4.0 cm sized lobulating solid mass at the posterolateral wall of the cervix, and both ovarian multiseptated cystic tumors were suspected to be mucinous borderline neoplasm (Fig. 1A). The serum levels of CA-125 (109.8 U/mL) and CA 19-9 (9,296 U/mL) were elevated and hemoglobin was markedly reduced (6.4 g/dL). The first biopsy of cervix was reported to be atypical endocervical glands. However, the re-biopsy of cervix was confirmed to be minimal deviation adenocarcinoma. The patient underwent radical hysterectomy with bilateral salpingooophorectomy, bilateral pelvic node dissection, paraaortic node sampling, omentectomy, and bladder biopsy.
Gossly, the cervix was markedly dilated and an infiltrating tumor mass was noted, measuring 5.5×4.0×4.5 cm (Fig. 1B). Microscopic findings of the cervix were compatible with minimal deviation adenocarcinoma (Fig. 1C). Histologically, both cystically dilated ovarian masses were reported to be metastatic MDA in mucinous cystadenoma with microscopic foci of SCTATs (Fig. 1D). The colonic polyps were confirmed to be hamartomatous polyp (Fig. 1E). The biopsy from the bladder flap was diagnosed as a direct extension of tumor cells. Immunohistochemically, in case 1, a reaction of rabbit polyclonal antibody to LKB1 (Novus Biologicals, Cambridge, UK) expression was not found in the atypical infiltrative glands (Fig. 1F). FIGO stage of case 1 was IIIA due to the extension into the lower third of vagina with pelvic lymph node metastasis.
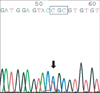
In case 1, DNA sequencing analysis was revealed a frame shift mutation of codon 132 (TGC to CGC) in exon 3 of the STK11/LKB1 gene (Fig. 2).
During the follow-up period, the patient underwent a colonoscopic polypectomy and was diagnosed with multiple hamartomatous polyps throughout the small intestine and entire colon. Her father had also been diagnosed with PJS and died due to colon cancer 10 years earlier.
The patient received combined chemo-radiation therapy with 3 cycles of irinotecan and cisplatin. Nevertheless, she complained of ever-intensifying lower abdominal pain due to the increasing masses in the pelvic cavity and the re-growing vaginal stump mass; she finally expired 1 year after the operation.
2. Case 2
A 35-year-old, single, virgin woman was referred to our hospital due to an abnormal finding of a Papanicolaou test, reported to be atypical glandular cells. Her past medical history was unremarkable except for melanin pigments on her digits and lips. Her parents and all of her siblings were alive and had no history of polyps or cancer in the gastrointestinal tract. She had undergone colonoscopic polypectomy at the age of 25 years and was diagnosed with a hamartomatous polyp.
A physical examination revealed a diffusely firm mass at the whole cervix. Pelvic MRI demonstrated a 4.4×3.3×2.2 cm sized mass at the whole cervix (Fig. 3A). The serum levels of CA-125 and CA 19-9 were within normal limits. The preoperative loop electrosurgical excision procedure (LEEP) of cervix was confirmed as minimal deviation adenocarcinoma. She also underwent radical hysterectomy with bilateral salpingooophorectomy and bilateral pelvic node dissection.
Grossly, a solid and infiltrating tumor mass at the whole cervix measures 4.5×3.0×2.5 cm (Fig. 3B). Microscopically, the cervical tumor was diagnosed as MDA (Fig. 3C) and scattered foci of tumorlets of SCTATs (Fig. 3D). Colonoscopic polypectomy confirmed the presence of a hamartomatous polyp (Fig. 3E).
Immunohistochemistry with the rabbit polyclonal antibody to LKB1 (Novus Biologicals) showed nuclear and cytoplasmic expression in atypical endocervical glands (Fig. 3F). FIGO of case 2 was IIA due to no tumor invasion of parametrium. She received combined chemo-radiotherapy with 3 cycles of paclitaxel and carboplatin regimen, and has no evidence of tumor recurrence for 5 years. In case 2, the STK11/LKB1 gene mutation was not found.
DISCUSSION
MDA is a very rare tumor and is composed of 1-3% of the uterine cervix. Approximately 10% of MDA is accompanied by PJS [3]. SCTATs is the most frequent ovarian tumor in patients with PJS [2]. Histologically, SCTATs share the features of granulose cells and a Sertoli cell tumor. SCTAT can produce excessive estrogen and progesterone; therefore, sometimes hyperestrogenic symptoms including irregular menstrual cycles, long lasting menstruation, and early puberty in girls have been mentioned [2]. SCTATs combined with PJS almost always present as benign behavior with scattered, bilateral, and very small or microscopic clusters [2]. However, SCTATs without PJS frequently have a larger tumor size and present with malignant clinical behavior.
Germ line mutations in the STK11/LKB1 gene were first reported in 1998 [5] and are responsible for PJS. Recently, germ line mutations in the STK11/LKB1 gene have been identified in 30-70% of patients with PJS [6].
The STK11/LKB1 gene is mapped on chromosome 19p 13.3 and is a tumor suppressor gene. A study of polyps accompanied by PJS reported that loss of heterozygosity of STK11/LKB1 causes loss of function of STK11/LKB1 [5]. Also promoter hypermethylation has been investigated due to inactivation of the STK11/LKB1 gene [7].
The STK11/LKB1 gene triggers cell cycle arrest through p21, which can be involved in apoptosis pathways, metabolism, cell polarity, and proliferation by activating the AMP-activated serine/threonine protein kinase (AMPK) pathway [8]. Furthermore, a number of studies have reported that one of the major downstream signaling pathways controlled by AMPK-LKB1 is the mammalian target-of-rapamycin pathway and this appears to be responsible for controlling protein synthesis, cell growth, and protecting against apoptosis during cellular stress [9].
Hearle et al. [10] have analyzed the incidence of cancer in 417 patients with PJS and detected 297 cases associated with the mutation of STK11/LKB1. They also found that the risk of cancer was similar in patients with PJS both with and without STK11/LKB1 mutation. Kuragaki et al. [3] have suggested that MDA with mutation of the STK11/LKB1 gene has a markedly poorer prognosis than MDA without mutation of the STK11/LKB1 gene. In our cases, case 1 had a frame shift mutation of the STK11/LKB1 gene with markedly aggressive clinical progression, and the patient died 1 year after diagnosis. However, case 2, without mutation of STK11/LKB1, has shown a better clinical course than that of case 1.
In conclusion, identification of a STK11/LKB1 mutation in patients with PJS can be used to anticipate and provide an early response to the presence of various tumors. Furthermore detection of a STK11/LKB1 gene mutation in cancer patients with PJS can help predict the prognosis and course of the disease, and can be used to improve clinical management.


 XML Download
XML Download