

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cytoreductive surgery followed by intermittent chemotherapy with taxane and a platinum chemotherapeutic agent is the standard treatment for ovarian cancer, and has an initial response rate of up to 80% [1,2]. The majority of patients, however, relapses after a median period of 18 months, and multi-drug resistance (MDR) to chemotherapeutic agents impedes successful chemotherapy of recurrent ovarian cancers [3]. This MDR phenomenon allows cancer cells to survive in the presence of structurally and functionally different chemotherapeutic agents, and is the main cause of therapeutic failure in ovarian cancers [4].
The most important mechanism responsible for the MDR phenotype is the overexpression of drug efflux transporter genes in cancer cells, such as P-glycoprotein (P-gp), an ATP-binding cassette (ABC) transporter [5,6]. P-gp transporter, encoded by the MDR1 gene, acts as a cell membrane pump extruding drugs to the extracellular space, thereby reducing drug accumulation in cells. As reported previously, various toxins and chemotherapeutic drugs, including taxanes and anthracyclines are MDR1 substrates [7].
Cyclooxygenase-2 (COX-2) is a key enzyme in converting arachidonic acid to prostaglandins, and it has been accepted as an unfavorable prognostic factor in various tumors, including ovarian cancer [8]. To overcome drug resistance by COX-2, COX-2 inhibitors have been studied in various cancers, such as leukemia, colon cancer, and hepatocellular carcinoma [9-12]. Selective COX-2 inhibitors have been recognized for their anticancer effects, in addition to their effects in treating rheumatoid arthritis and managing pain [13]. Moreover, selective COX-2 inhibitors sensitize MDR cancer cells to chemotherapeutic drugs in either COX-2-dependent or COX-2-independent mechanisms [14].
The aim of the present study was to demonstrate the usefulness of selective COX inhibitors in promoting paclitaxel-induced apoptosis in taxane-resistant ovarian cancer cells, and to understand the mechanisms involved in the selective COX inhibitors suppressing MDR1 gene expression.
MATERIALS AND METHODS
1. Reagents and antibodies
NS-398 and SC-560, selective COX inhibitors, were purchased from Cayman Chemical Co. (Ann Arbor, MI, USA). Paclitaxel was purchased from the Cheil General Hospital Pharmacy (Seoul, Korea). LY294002, a selective PI3K inhibitor, was purchased from Cell Signaling Technology (Beverly, MA, USA). Prostaglandin E2 (PGE2) was purchased from Sigma-Aldrich (St Louis, MO, USA). Antibodies against COX-1, COX-2, and cleaved poly ADP ribose polymerase (PARP) were supplied by Cell Signaling Technology (Beverly, MA, USA). The anti-MDR1 antibody was obtained from Abcam (Cambridge, UK). The anti-actin antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-rabbit and anti-mouse horseradish peroxidase-conjugated secondary antibodies were obtained from Cell Signaling Technology.
2. Cell culture
Human ovarian carcinoma cell lines SKOV3ip1, HeyA8 (taxane-sensitive), SKOV3ip2-TR, HeyA8-MDR (taxane-resistant) were provided by Dr. AK Sood (Texas MD Anderson Cancer Center, TX, USA). SKOV3ip1 and HeyA8 cells were grown in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and 0.5% gentamicin. SKOV3ip2-TR and HeyA8-MDR cells were grown in RPMI 1640 supplemented with 10% FBS and 0.5% gentamicin, and with 300 ng/mL paclitaxel.
3. Immunoblot analysis
Cells (5×105) were lysed in a lysis buffer (10 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulphate [SDS]), proteins were resolved by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred onto nitrocellulose membranes (Millipore, Bedford, MA, USA). Membranes were blocked with 5% skim milk in tris-buffered saline with tween 20 (TBS-T) buffer (10 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.1% Tween-20) for 1 hour and then incubated with relevant antibodies for 18 hours at 4℃. Membranes were washed with TBS-T buffer and incubated for 1 hour at room temperature (RT) with horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG at a dilution of 1:5,000. Protein bands were visualized using enhanced chemiluminescence (Amersham-Pharmacia, Piscataway, NJ, USA).
4. MTT assay
Cells (3×103) were seeded in 96-well microplates treated with COX inhibitors, PGE2, or paclitaxel as indicated. At 72 hours after incubation, 100 µL/well of 2 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution was added to the microplates. Two hours after MTT treatment, the medium was removed and formazan crystals were dissolved by adding 100 µL dimethylsulfoxide per well. Cell viability was analyzed by measuring the absorbance at 590 nm using an enzyme-linked immunosorbent assay reader.
5. Total RNA isolation and reverse transcription polymerase chain reaction
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). One microgram of the isolated total RNA was converted to cDNA using M-MLV reverse transcriptase (Promega, Madison, WI, USA). AvPCR primer set specific for each gene was used for amplification. PCR products were resolved on a 2.0% agarose gel and visualized by ethidium bromide staining.
6. Cell cycle analysis
Cells (5×105) were harvested and fixed with 70% (w/v) ice-cold ethanol at 4℃ for 1 hour. Fixed cells were washed twice with phosphate-buffered saline (PBS) and stained with PBS containing 50 µg/mL propidium iodide (PI) and 100 µg/mL Ribonuclease A (RNase A). Following incubation for 15 minutes in the dark at RT, the number of cells in each cell cycle stage was analyzed using FACSCalibur Flow Cytometry (BD Bioscience, Franklin Lakes, NJ, USA) and CellQuest software (BD Bioscience).
RESULTS
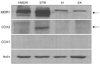
1. P-gp is upregulated in taxane-resistant ovarian cancer cells
To investigate whether taxane resistance is associated with increased expression of MDR1/P-gp, COX-1, or COX-2, we examined their expression in ovarian cancer cell lines (HeyA8-MDR, SKOV3ip2-TR, HeyA8, and SKOV3ip1). As expected, the taxane-resistant ovarian cancer cell lines (HeyA8-MDR and SKOV3ip2-TR) expressed more MDR1/P-gp than the taxane-sensitive ovarian cancer cell lines (HeyA8 and SKOV3ip1) (Fig. 1). COX expression, however, was inconsistent. COX-1 was not expressed in all cell lines, and COX-2 was only expressed in SKOV3ip2-TR, a taxane-resistant cell line (Fig. 1).
2. Selective COX inhibitors significantly enhance the cytotoxic effects of paclitaxel in taxane-resistant ovarian cancer cells
To determine the cytotoxic effect of COX inhibitors in paclitaxel-resistant ovarian cancer cells, the viability of cells treated with COX inhibitors was measured using an MTT assay. The results show that selective COX inhibitors enhanced paclitaxel cytotoxiticy in the taxane-resistant ovarian cancer cell lines (HeyA8-MDR and SKOV3ip2-TR) (Fig. 2). Subsequently, paclitaxel-resistant ovarian cancer cells were treated with paclitaxel (100 nM) combined with a selective inhibitor for COX-1 (SC560) or COX-2 (NS398) at various concentrations. SC560 and NS398 significantly enhanced the cytotoxic effects of paclitaxel at 20 and 100 µM, respectively, in SKOV3ip2-TR cells. In HeyA8-MDR cells, SC560 significantly increased the cytotoxic effect of paclitaxel at 20 µM.
3. Combined treatment with paclitaxel and a selective COX inhibitor promotes apoptosis in taxane-resistant ovarian cancer cell lines.
To investigate whether paclitaxel and COX inhibitors could enhance apoptosis in taxane-resistant ovarian cancer cells, we analyzed the distribution of HeyA8-MDR and SKOV3ip2-TR cells in various cell cycle stages when treated with paclitaxel (100 nM) alone or combined with SC560 (20 µM) or NS398 (100 µM) (Fig. 3A, B). Combined treatment with paclitaxel and SC560 or NS398 increased the sub-G1 fraction. Furthermore, immunoblot analysis for PARP cleavage revealed that combined treatment with paclitaxel and selective COX inhibitors increased the cytotoxic effect in taxane-resistant ovarian cancer cells (Fig. 3C). These results indicate that selective COX inhibitors increase apoptosis in taxane-resistant ovarian cancer cells treated with paclitaxel.
4. Selective COX inhibitors promote apoptosis of taxane-resistant ovarian cancer cells in a prostaglandin-independent manner
To determine whether enhanced apoptosis of taxane-resistant ovarian cancer cells treated with paclitaxel and a selective COX inhibitor was mediated by prostaglandins, we added PGE2 (10 µM) to taxane-resistant ovarian cells treated with paclitaxel and COX inhibitors (Fig. 4). Adding PGE2 did not change the effects of COX inhibitors in taxane-resistant ovarian cancer cells. These results indicate that selective COX inhibitors may overcome paclitaxel resistance by ovarian cancer cells in a prostaglandin-independent manner.
5. Selective COX inhibitors suppress MDR1 and P-gp expression in HeyA8-MDR and SKOV3ip2-TR cells
To clarify the precise mechanisms by which selective COX inhibitors inhibit the growth of paclitaxel-resistant ovarian cancer cells, MDR gene expression was analyzed in ovarian cancer cells treated with a COX inhibitor. Combined treatment with paclitaxel and COX inhibitors reduced expression of the MDR1 gene (Fig. 5A) and P-gp protein (Fig. 5B). These findings suggest that selective COX inhibitors suppressed MDR1/P-gp expression in taxane-resistant ovarian cancer cells.
DISCUSSION
The aim of this study was to investigate whether the selective COX inhibitors promote paclitaxel-induced cytotoxicity in taxane-resistant ovarian cancer cells by suppressing MDR1/P-gp expression, and to clarify whether these effects depend on the COX pathway. We found that the combined treatment with paclitaxel and a selective COX inhibitor promoted cytotoxicity in taxane-resistant ovarian cancers by suppressing MDR1 gene and P-gp protein expression (Fig. 5). Our results correspond well with previous studies that reported that COX inhibitors significantly enhanced the cytotoxic effects of chemotherapeutic agents in drug-resistant cancer cells by inhibiting P-gp expression [11,12,15-17].
In this study, we documented that P-gp is overexpressed in the taxane-resistant ovarian cancer cell lines, HeyA8-MDR and SKOV3ip2-TR, but not in the taxane-sensitive cell lines, HeyA8 and SKOV3ip1. COX expression, however, was inconsistent among these cell lines. COX-2 was only expressed in one taxane-resistant cell line, SKOV3ip2-TR (Fig. 1). Because taxane-resistant ovarian cancer cells did not express significantly more COX than taxane-sensitive ovarian cancer cells, the inhibitory effect of selective COX inhibitors on MDR1/P-gp expression may not be mediated through the COX pathway. Thus, COX inhibitors may increase intracellular accumulation of paclitaxel and subsequently enhance cytotoxicity by directly suppressing P-gp expression, without depending on the COX pathway.
We showed that adding the end product of COX pathway, PGE2, did not reverse the inhibitory effects of selective COX inhibitors on paclitaxel-resistant ovarian cancer cell growth (Fig. 4), which further suggests that the cytotoxic effects of COX inhibitors are COX- and PGE2-independent. These findings are consistent well with previous studies [12,17,18]. Xia et al. [17] reported that a selective COX-2 inhibitor, celecoxib, enhances the drug sensitivity of human breast cancer cells. Ye et al. [12] showed that indomethacin and a selective COX-2 inhibitor enhanced doxorubicin cytotoxicity in human hepatocellular carcinoma cells, by inhibiting P-gp expression in a COX-2-independent manner. However, Roy et al. [11] reports results that are in contrast ours. They suggested that selective COX inhibitors regulate MDR1 expression and potentiate the effects of doxorubicin in human hepatocellular carcinoma cells by altering the activity of ABC proteins in a COX-2 dependent manner. They concluded that the cytotoxicity of COX-2 inhibitors is mediated by down-regulating MDR1 in a PGE2- or COX-dependent manner. Interestingly, our study showed that taxane-resistant ovarian cancer cells did not consistently express more COX-1 or COX-2 than taxane-sensitive ovarian cancer cells. Further, adding PGE2, the end product of the COX pathway, did not reverse the effects of selective COX inhibitors. Xia et al. [17] also suggested that the effects of selective COX inhibitors on P-gp are COX-2-independent but closely related to hypermethylation of the MDR1 promoter. Further studies are required to elucidate the precise mechanisms involved in chemosensitization by COX inhibitors.
With taxane-sensitive ovarian cancer cell line, SKOV-3, Li et al. [19,20] reported that combined treatment of COX inhibitors with taxol has effects on angiogenesis and apoptosis in human ovarian cancer xenografts. They report that COX inhibitor enhances the anti-angiogenic and pro-apoptotic effects of taxol in taxane-sensitive ovarian cancer in vivo, thus they suggest combined treatment of COX inhibitors with taxol in ovarian cancer therapy. Our studies with taxane-sensitive ovarian cell lines were also consistent with theirs (data not shown). The synergistic effects of paclitaxel and COX inhibitor were also seen in taxane-sensitive HeyA8 and SKOV3ip1 cell lines. These findings suggest that COX inhibitor can be a potent therapeutic tools not only as a drug sensitizer, but also as an anti-angiogenic and pro-apoptotic agents.
In conclusion, our data imply that selective COX inhibitors significantly increase paclitaxel-induced cell death in taxane-resistant ovarian cancer cells in a prostaglandin- and COX-independent manner. These findings suggest that combined treatment with selective COX inhibitors and paclitaxel could be a potent therapeutic tool to promote paclitaxel sensitization of taxane-resistant ovarian cancers by suppressing MDR1/P-gp expression, which is responsible for efflux of chemotherapeutic agents. Thus, selective COX inhibitors could be good chemosensitizers to improve chemotherapy.




 XML Download
XML Download