

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Ovarian cancer is a very lethal type of gynecologic cancer with over 100,000 people dying every year from this disease around the world [1]. Among patients diagnosed with ovarian cancer, 75% of the cases are initially detected at the disseminated (stage III/IV) form of the disease and the 5-year survival rate is less than 25%. Primary treatment for ovarian cancer is cytoreductive surgery followed by combination chemotherapy. More than 70% of patients typically respond to primary treatment [2]. However, more than 85% of those responders will relapse despite surgical debulking, followed by platinum (e.g., cisplatin or carboplatin) and taxane regimens [2]. Despite the initial response, relapse is mainly caused by resistance to chemotherapeutic agents. At the time of recurrence, additional therapy options are very limited because secondary chemotherapeutic agents cause more serious side effects and stronger toxicity. Reducing resistance to chemotherapeutic agents may greatly decrease the mortality of ovarian cancer and relapse of this devastating disease.
The primary role of transcription is to regulate the expression of specific target genes. Epigenetic mechanisms like histone modification and DNA methylation have been known to play a key role in the regulation of gene transcription. Variation of epigenetic regulation is one of the known causes of abnormal gene expression in a malignant tumor resistant to chemotherapy. Specially, acetylation of DNA-bound core histones is associated with up-regulation of transcription and is regulated by two opposing classes of enzymes, histone acetyltransferases (HATs) and histone deacetylases (HDACs). Acetylation of DNA sequence-specific transcription factors also regulates gene transcription. HDACs are emerging as critical regulators of cell growth, differentiation, and apoptosis. Recently, several HDAC inhibitors were studied and demonstrated considerable anti-tumor effect in various types of cancers including colonic carcinoma, pancreatic cancer, and hepatoma [3-5]. For example, a phase I clinical trial of suberoylanilide hydroxamic acid (SAHA), an HDAC inhibitor that suppresses the activity of HDAC class I and II, demonstrated anti-tumor activity in solid and hematologic tumors [6]. In addition, several clinical trials of HDAC inhibitors that included desipeptide, MS-275, LAQ-824, LBH-589, and MGCD 0103, were reported to have a strong anti-tumor activity in various types of cancer [7-11].
For ovarian cancer, if resistance to chemotherapeutic agents is removed, mortality and morbidity would decrease and the use of more toxic secondary agents may be reduced. The overexpression of HDAC 1, 2, and 3 has previously been reported in ovarian cancer tissues [12]. We hypothesized that the mechanism of resistance to chemotherapeutic agents is associated with overexpressed HDACs in ovarian cancer cells.
In this study, we investigated the relationship between cisplatin resistance and HDAC overexpression in two epithelial ovarian cancer cell lines, SKOV3 and OVCAR3. HDAC isoforms associated with cisplatin resistance in each cell line were compared.
MATERIALS AND METHODS
1. Materials
Components for cell culture media were purchased from Life Technologies (Gaithersburg, MD, USA) unless otherwise noted. The SKOV-3 and OVCAR3 human epithelial ovarian cancer cell lines were obtained from the Korean Cell Line Bank (Seoul, Korea). The Taq DNA polymerase PCR system was purchased from Takara Bio Inc. (Shiga, Japan). Polyclonal antibodies to HDAC 1-4 and acetylated histone 3 were purchased from Cell Signaling (Danvers, MA, USA) and horseradish-peroxidase conjugated secondary antibodies were obtained from Jackson ImmunoResearch Laboratory (West Grove, PA, USA). From Sigma-Aldrich (St. Louis, MO, USA) we obtained mouse monoclonal antibody to β-actin, complete protease inhibitor cocktail, and cisplatin. AC 1-4 expression vectors were kindly provided by Dr. Hyun Kook from the Department of Pharmacology, Chonnam University Medical School, Gwangju, Korea. All biotechnology-grade chemicals were purchased from Amresco Inc. (Solon, OH, USA).
2. Cell culture
OVCAR3 and SKOV3 cells were cultured in RPMI 1640 media supplemented with 100 units/mL penicillin, 100 µg/mL streptomycin, and 10% fetal bovine serum (FBS) at 37℃ in a 5% CO2 incubator. Cells exposed to cisplatin were plated at 3×105 cells per well in a 6-well culture dish for 24 hours before use. Cells were treated with various concentrations of cisplatin and were incubated for a specific length of time.
3. Cytotoxicity assay
Cell viability was examined using the colorimetric cell counting kit-8 (CCK-8) assay (Dojindo Lab., Kumamoto, Japan) according to manufacturer instructions. Cells were seeded at a density of 3×103 per well in a 96-well plate and were cultured as described above. Cells were then treated with 15 µg/mL cisplatin for 0, 15, 24, 36, and 48 hours. The amount of formazan dye generated by cellular dehydrogenase activity was measured by absorbance at 450 nm using an ELISA reader (Molecular Devices Corporation, Sunnyvale, CA, USA). Absorbance values were converted to percentages for comparison with untreated control. We used IC50 Calculate software (LOGIT method) for calculating the 50% inhibitory concentration (IC50).
4. Transfection
Cells were plated 24 hours prior to transfection at a density of 2×105 cells per 35 mm culture dish. Cells were transiently transfected using Lipofectamine LTX (Invitrogen, Carlsbad, CA, USA) and 2.5 µg of each HDAC isoform expression vector or mock vector as a control. Twenty-four hours after transfection, the cells were treated with cisplatin for the indicated length of time.
5. Nuclear protein extraction for the detection of histone 3 (H3) acetylation
Cells were harvested and nuclear proteins were isolated using Cellytic Nuclear TM Extraction Kit (Sigma-Aldrich) according to manufacturer instructions. Nuclear extract protein levels were measured and the extracts were stored at -20℃ until use.
6. Western blot analysis
Cells were washed twice with phosphate-buffered saline (PBS) on ice and were disrupted with RIPA lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP-40, 0.25% Na-deoxychalate, 1 mM EDTA, 1 mM NaVO4, 2 mM NaF, and complete protease inhibitor cocktail). The extracts were homogenized by sonication and centrifuged at 13,000 rpm for 20 minutes at 4℃. Protein concentration was measured using the BCA Protein Assay (Pierce, Rockford, IL, USA). Protein samples (30 µg per lane) were resolved on SDS-polyacrylamide gel and electrophoretically transferred to nitrocellulose membranes (GE Healthcare Biosciences, Uppsala, Sweden). After blocking with 5% skim milk in TBS-T buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.5% Tween-20), the membranes were probed with primary antibodies that correspond to target proteins. Membranes were then washed three times with TBS-T buffer followed by incubation with horse radish-conjugated secondary antibody (Jackson ImmunoResearch Lab). The reaction was detected with enhanced chemiluminescence (ECL; GE Healthcare Biosciences) and exposed to X-ray film. To normalize for protein loading, the membranes were washed with stripping solution (62.5 mM Tris-HCl [pH 6.8], 100 mM β-mercaptoethanol, 2% SDS) at 55℃ and reprobed with a monoclonal antibody to β-actin.
7. Total RNA extraction and RT-PCR analysis
Cells were washed twice with cold PBS and total RNA was extracted using TRIzol (Invitrogen) according to manufacturer instructions. The RNA samples were quantified, aliquoted, and stored at -80℃ until use. Approximately 1 µg of total RNA was used to synthesize first-stranded cDNA using the SuperScript II First-Strand System (Invitrogen). The PCR was performed for 30 cycles of denaturation at 94℃ for 1 minute, annealing at 55℃ for 45 seconds, extension at 72℃ for 45 seconds. After completing 30 cycles, a final extension cycle of 72℃ for 2 minutes was carried out. Table 1 illustrates a pair of DNA oligonucleotides designed with PrimerDesigner program, based on the cDNA sequences of each human HDAC isoform and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in the GenBank database. The PCR products were separated on a 1% agarose gel and images were captured using an ultraviolet plate.
8. Statistical analysis
Cell survival rates for the control and test groups after treatment with cisplatin were presented as average±standard deviation. The statistical analysis was performed using the Mann-Whitney U-test employing the SPSS ver. 12.0 (SPSS Inc., Chicago, IL, USA). A value of p<0.05 was considered statistically significant.
RESULTS
1. Cytotoxicity test
After treatment with 15 µg/mL cisplatin, the survival rate of SKOV3 and OVCAR3 cells decreased with time and both groups reached IC50 between 15 and 24 hours (Fig. 1). Although there were no significant differences in the survival rates of SKOV3 and OVCAR3 cells until 15 hours, SKOV3 cells did show more survival compared with OVCAR3. It shows that SKOV3 has more resistance than OVCAR3 to cisplatin.
2. Histone acetylation and deacetylation by cisplatin
After treatment of SKOV3 cells with 15 µg/mL cisplatin, nuclear protein was separated and the level of expression of acetylated H3 was measured. The level of H3 expression was increased, especially around 24 hours (Fig. 2). After treatment with cisplatin for 24 hours, we compared the protein expression levels of HDAC 1, 2, 3, and 4. No meaningful changes in the expression level were observed for HDAC 2 and 3 despite an increase in cisplatin exposure. At the concentration of 15 µg/mL cisplatin, HDAC 1 expression significantly decreased. HDAC 4 expression markedly decreased from the concentration of 4 µg/mL cisplatin and was nearly undetected above 4 µg/mL cisplatin (Fig. 3). From these results, it may be estimated that HDAC 1-3 are associated primarily with histone deacetylation in SKOV3 cells.
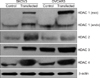
4. Relationship between overexpression of HDAC proteins and resistance to cisplatin
After overexpressing HDACs in SKOV3 and OVCAR3 cells treated with 15 µg/mL cisplatin, we compared differences in cell viability among the HDAC isoforms. In the case of SKOV3, the IC50 of the control group was observed at 24 hours. The viability of cells overexpressing HDAC 1 and 3 increased up to 15% and 13%, respectively, compared with control cells at 24 hours (p<0.05) (Fig. 6). In the case of OVCAR3, the viability of cells overexpressing HDAC 2 and 4 increased up to 23% and 20%, respectively, compared with control at cells at 24 hours (p<0.05) (Fig. 7). These results suggest that increased expression of various types of HDAC increased cisplatin resistance for both cell lines. In addition, the resistance conferred by different HDAC isoforms depended on the type of ovarian cancer cell line that was transfected.
DISCUSSION
Gene expression is not determined only by the DNA code but also by epigenetic modifications. Epigenetic modifications can be mitotically inherited for more than one generation [13]. However, epigenetic mechanisms can be modified by chemical agents and not by DNA sequence alterations. Despite extensive research into the biological functions of HDACs in various types of cancer, there are few reports on the relationship of HDACs and anti-cancer drug resistance. Previous studies from our institution demonstrated that HDACs 1-3 were elevated in ovarian cancer tissue at both transcriptional and translational levels. It is suspected that overexpressed HDACs may play a significant role in ovarian carcinogenesis. Recently, Muscolini et al. [14] showed that an HDAC inhibitor induces apoptosis in cisplatin-resistant ovarian cancer cells.
In this study, we showed that overexpression of HDACs is associated with resistance to cisplatin, a chemotherapeutic agent used for treating ovarian cancer. Furthermore, we demonstrated that the specific HDAC isoform associated with resistance to cisplatin was different according to the specific type of ovarian cancer cell line. Our study may provide the scientific basis for the clinical application of HDAC inhibitors in cisplatin-resistant ovarian cancer.
The epigenetic regulation of genes can be divided into two different mechanisms: structural modification of chromatin, and covalent modification of histone protein or DNA methylation. These mechanisms are proven in fields of research such as; cancer biology [15], viral latency [16], somatic gene therapy [17], genomic imprinting [18], etc. The human genome is packaged as chromatin which consists of DNA, histone and non-histone protein. Chromatin remodeling is important in epigenetic regulation of gene expression. The tail regions of both H3 and H4 are covalently modified where the ε-amino group of lysine in the N-terminal region of histone acts as a substrate for HDAC and HAT. The histone tails protruded from the chromatin polymer and are very sensitive to protease. This modification is commonly called the histone code or epigenetic code and may be controlled by HAT, HDAC, histone methyltransferase (HMT), and histone kinase (HK).
Hypoacetylated histone containing positive charges within the nucleic acid may be combined with a phosphate backbone to cause transcriptional silencing of the chromatin. At that time, acetylation of histone may neutralize positive charges, changing the structure of the chromatin and making it easier to access transcription factors, the transcription regulatory complex and RNA polymerase, among others. In contrast, histone deacetylation may restore positive charges of histone lysine, and turn the chromatin to a very supercoiled state and cause genetic silencing [19]. HDAC and HAT can affect transcription by selectively acetylating or deacetylating the ε-amino group at the amino terminus in core histone.
HDACs modify core histones and participate in large regulatory complexes that both suppress and enhance transcription. There are 18 HDACs identified in humans which can be classified into 4 categories on the basis of homology to yeast HDACs and phylogenic analysis [20,21]. Class I HDACs are homologous to yeast RpD3 and are located primarily in the nucleus. Class I HDACs include HDAC 1-3 and 8. As an exception, HDAC 3 moves between the cytoplasm and the nucleus. Class I HDACs appear to play a primary role in cell survival and proliferation and are mainly associated with transcriptional repressors and cofactors. HDAC 1 and 3 are known to associate with transcription factor and hypoxia inducible factor (HIF)-1α, as well as affect angiogenesis [22]. HDAC 2 can modulate transcriptional activity through interaction with p53. HDAC 4 serves as a nuclear co-repressor involved in regulating bone and muscle development, and regulates neuronal survival in retinas [23]. Class II HDACs are homologous to yeast Hda1 and include HDAC 4-7, 9, and 10. Class II HDACs exist both in the nucleus and the cytoplasm. Class II HDACs have tissue-specific roles. HDAC 6 and 10 have two catalytic domains, C-terminal and N-terminal. Class III HDACs are homologous to yeast Sir 2. Class III HDACs are sirtuins 1-7 which have an absolute requirement for NAD+, while class I, II, and IV HDACs are zinc dependent. Class IV HDAC (HDAC 11) has both features of class I and class II. Class I and II HDACs are sensitive to the already known HDAC inhibitors [24]. However, the functions of most class III HDACs and class IV HDAC (HDAC 11) remains to be elucidated.
It has already been proven that epigenetic alteration plays an important role in carcinogenesis, tumor growth, and progression. Aberration of DNA methylation and histone deacetylation may be a significant cause of resistance to chemotherapeutic agents. This aberration occurs in the course of apoptosis and differentiation. The resensitization of tumor cells to chemotherapeutic agents could be achieved by reforming this abnormal modification. Thus, as epigenetic chemotherapy and conventional chemotherapy are combined, tumor cells that were resistant to chemotherapeutic agents are resensitized and would once again respond to first-line chemotherapeutic agents such as taxane and cisplatin.
We demonstrated that cancer cells that overexpress HDACs show resistance to cisplatin. HDAC 1 and 3 were involved in SKOV3 resistance to cisplatin, while HDAC 2 and 4 were involved in OVCAR3 resistance to cisplatin. The difference of HDAC isoforms may be due to the features of cell lines themselves or to the different mechanism of action for each HDAC isoform. In SKOV3, the p53 gene, which is important in the process of carcinogenesis, is deleted while in OVCAR3 it is mutated. The p53 gene activates proapoptotic proteins like Bax, Bam, Puma, and Noxa through a variety of pathways and suppresses antiapoptotic proteins like Bcl2. Thus, p53 plays an important role in inducing apoptosis and cell cycle arrest [25]. HDAC 1 suppresses p53-mediated apoptosis by suppressing the activity of p53 on the Bax promoter while HDAC 2 suppresses the DNA-binding activity of p53 [26]. Moreover, HDAC 3 and 4 are implicated in transcription suppression, with HDAC 3 suppressing the p53-p21 pathway and HDAC 4 causing posttranslational modification (acetylation or methylation of the C-terminal lysine of p53 protein) [27,28]. In addition, HDACs can also take part in apoptosis through other pathways, regardless of p53. However, the function of HDACs on p53 still remains unclear. Based on this study, HDAC 1 and 3 may provide resistance to apoptosis independent of p53 via another pathway, and HDAC 2 and 4 may increase resistance via p53 mutation or another p53-mediated pathway in addition to another p53 non-mediated pathway.
In this study, it was identified that when HDAC 1, 2, 3, and 4 were overexpressed in SKOV3 and OVCAR3 ovarian cancer cell lines, resistance to cisplatin was conveyed when compared with a control group not containing overexpressed HDACs. HDAC 1 and 3 in SKOV3, and HDAC 2 and 4 in OVCAR3 were strongly associated with cisplatin resistance. These results suggest that the HDAC isoforms resistant to cisplatin in the two cell lines are different from each other. If we could find the specific HDAC isoforms associated with cisplatin resistance in ovarian carcinoma, combination therapy with HDAC inhibitors may magnify the effects of primary chemotherapeutic agents in the treatment of ovarian carcinoma while reducing the toxicity of secondary anti-cancer drugs.







 XML Download
XML Download