

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Since Lynch first described two major cancer families in 1966 [1], Lynch syndrome or hereditary nonpolyposis colorectal cancer (HNPCC) has been reported to occur in a variety of extracolonic sites. He described gynecologic malignancies including 15 uterine, four ovarian, and one cervical cancer in his original publication of major cancer families. Autosomal germline mutations of DNA mismatch repair genes are thought to give rise to Lynch syndrome. The most commonly affected genes are MLH1 (40-45% of cases), MSH2 (40-45%), MSH6 (5-10%), and PMS2 (<5%) [2-7]. These genetic defects in the DNA mismatch repair system result in replication errors in repetitive DNA segments, known as microsatellite instability (MSI), and the absence of protein expression in the tumor. Currently, the diagnosis of Lynch syndrome is diagnosed based on family history, immunohistochemistry (IHC), MSI, and gene sequencing chromatogram (revised Amsterdam, 1998 [8] or Bethesda, 2002 [9] criteria).
Endometrial and ovarian cancers have been reported to occur in 60% and 12% of patients with Lynch syndrome, respectively [10,11]. Synchronous (or multiple primary) gynecologic tumors are uncommon. Among such cases, a previous study estimated the frequency of Lynch syndrome to be 7-14% according to clinical and molecular diagnostic algorithms [12,13].
Frequency of Lynch syndrome in Korean endometrial cancer patients has been reported to be 3.5% (4/113) [14] and 11.2% (18/161) [15], but there are no reports describing synchronous gynecologic tumors. Therefore, we undertook the present study to evaluate the frequency of Lynch syndrome in Korean women with synchronous gynecologic tumors including endometrial and ovarian cancers based on family history and IHC.
MATERIALS AND METHODS
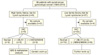
Our study sample was identified through a search of medical records. Between 1995 and 2010, we identified 36 women with synchronous cancers (endometrial and ovarian, n=36) patients who underwent treatment at Samsung Medical Center. Clinical and pathologic data were obtained. Inclusion criteria were: 1) synchronous gynecologic malignancy (combination of two types among endometrial and ovarian cancer); and 2) family history of cancer confirmed by medical records and telephone interview. Patients were categorized into risk groups based on reported family history: high risk (family history of cancer), and low risk (no relevant family history). Family history was evaluated by medical records about site of cancer, age of diagnosis and relation from patient. But when medical records were incomplete, telephone interviews were done.
National Cancer Institute's Surveillance, Epidemiology, and End Results (SEER) defines synchronous tumor as diagnosed two sites tumors within 2 months of each other. Our institution followed the criteria [16]. A gynecologic pathologist (SYS) reviewed the hematoxylin and eosin-stained slides from all tumors to confirm the diagnosis of synchronous primary cancers based on characteristic findings (no tumor between two sites, no metastasis or spread of disease from one site to another).
All independent gynecologic synchronous primary tumors confirmed based on the criteria by Scully et al. [17] in 1998 were included. Metastasis and spread from other sites were excluded. Sixty two paraffin-embedded tumor blocks were available for 32 patients.
IHC analysis was performed on all tumor specimens to determine the protein expression of MSH2, MSH6, and MLH1. As shown in Fig. 1, staining was scored based on intensity and proportion (negative or 0: intensity undetectable or minimal, proportion <5%; weak or 1+: intensity mild, proportion 5-30%; strong or 2+: intensity moderate to marked, proportion 30-99%).
For IHC analysis, monoclonal antibodies against MSH2 (Leica microsystems, Wetzlar, Germany), MSH6 (BD Transduction Laboratories, Lexington, KY, USA), and MLH1 (Leica microsystems) were used. Immunostaining was performed with Bond-Max (Leica microsystems) according to the manufacturer's instructions. The normal staining patterns for MSH2, MSH6, and MLH1 are nuclear. Loss of expression of MSH2, MSH6, and MLH1 in cancer cells was demonstrated by the total absence of nuclear staining in the tumor. Adjacent normal stroma or infiltrating lymphocytes served as an internal positive control for each case. Nuclear staining of the tumor was scored as either 2 (strong), 1 (weak), or 0 (negative) compared with the corresponding internal control.
Once all data were collected, the likelihood of each patient having Lynch syndrome/HNPCC was determined based on a combination of clinical and IHC criteria that included a family history consistent with Lynch syndrome and MLH1, MSH2, and MSH6 protein expression (both tumor sites negative or negative staining for more than two of these proteins).
RESULTS
Demographic characteristics for the 36 patients are listed in Table 1. The median age at diagnosis was 48 years (range, 26 to 71 years). Based on family history, 9 patients (25%) were considered high risk, and 27 (75%) were considered low risk. Fifty percent (18/36) were both endometrioid in histology (Table 2). The remaining were serous, mucinous, and mixed endometrioid and mucinous adenocarcinoma. The two most common ovarian histologies were endometrioid (n=18), and mucinous (n=8), followed by serous (n=5), transitional (n=2), clear cell (n=2), and undifferentiated (n=2). Among the 32 cases whose tumors were available, 15 patients (47%) had loss of at least one of the following: MSH2, MSH6, or MLH1 (Tables 3 and 4).
Six patients showed loss of MLH1 and the concordance rate of both tumor sites was 67% (4/6). Two patients showed weak MLH1 staining. Four patients showed results with both sites negative for MLH1 (4/6; cases 1-4 in Table 4). Among them case 2 is a high risk group.
Five patients were negative for MSH2 and 8 were negative for MSH6 at any site. Three patients showed weak staining for MSH2 and 5 showed weak staining for MSH6. Of the three with weak MSH2 staining, two showed weak MSH6 staining and one was negative for MSH6. Because of the high positive relationship between MSH2 and MSH6 staining and Lynch syndrome, we estimated that cases with both sites negative for MSH2 and MSH6 or negative for both proteins at any site were associated with Lynch syndrome (4/9 cases 5, 7, 8, 9 in Table 4). Among them cases 7 and 8 are high risk group. Case 2, 7, and 8 had a family history and valid IHC results associated with Lynch syndrome risk.
In addition, we reserved 9 patients with low family history risk but appropriate staining results (negative MLH1, n=3 or negative MSH2 and MSH6, n=6) for further workup and genetic tests.
The frequency of Lynch syndrome was estimated as 9% (3/32) among Korean women with synchronous gynecologic tumors based on a combination of family history and IHC results (loss of MSH2, MSH6, and MLH1 expression).
Fig. 2 provides a summary of our findings. The majority of patients (27/36, 75%) did not have family histories of cancer. Among these low-risk women, we were able to obtain 45 tumor specimens. A few of these women had IHC findings suspicious of a MMR defect: three patients showed negative staining of MLH1 in both sites, one patient was MSH2 and MSH6 negative, and another was evaluated as MSH6 negative at both sites.
DISCUSSION
Among Lynch related cancers, endometrial cancer is much riskier than colorectal cancer in terms of lifetime incidence [18]. Ovarian cancer related to Lynch syndrome was reported to account for 3-14% of ovarian cancer patients. Considering gynecologic malignancy surgery principles, hysterectomy and oophorectomy can be performed in cases of either endometrial or ovary cancer. The uterus or the ovary can be saved for early stage and young women. Especially, ovary can be saved in early stage endometrial cancer. Therefore, Lynch syndrome-related gynecologic malignancy takes a major part of it.
Synchronous primary cancers of the endometrium and ovary have been identified in 7-29% of young women diagnosed with endometrial cancer [19,20]. Although the etiology of these cancers has not been clearly determined, familial cancer syndromes often account for patients diagnosed with multiple cancers or patients diagnosed with cancer at a young age. Lynch syndrome (HNPCC) is an autosomal-dominant cancer predisposition syndrome that increases risk for multiple cancers.
Our study is the first report of the frequency of Lynch syndrome in synchronous gynecologic malignancy patients in Korea based on clinical and IHC criteria (MLH1, MSH2, and MSH6 protein expression). Our diagnosis was based on several clinical and IHC criteria. The IHC criteria were both sites negative for MLH1 or MSH2/MSH6, or negativity for both MSH2/6 and MLH1 at any site. According to our algorithm, the frequency of Lynch syndrome was estimated as 9% (3/36) (case 2, 7, 8 in Table 4) among Korean women with synchronous gynecologic tumors.
The frequency of Lynch syndrome in synchronous gynecologic tumors (combination of endometrium and ovary) has been reported to be 7% (7/102) [12] and 14% (3/22) [13]: however, more expensive and more precise gene tests were used (e.g., MSI or gene sequencing) and Asians represented only 2-14% of the study samples (2/102 and 3/22). Therefore, our study provides pilot results suggestive of a prospective study. Based on our findings, we can better counsel patients at risk of this syndrome and their relatives regarding an expensive gene test for prevention of other cancers.
Our recommendations for future study are: 1) multicenter investigation of synchronous gynecologic malignancy; 2) cost effective and appropriate selection of genetic testing for patients; 3) efficient MLH1, MSH2, and MSH6 IHC analysis during endometrial or cervical biopsy for patients with family history of cancer; 4) cooperation and close follow-up with a gastrointestinal physician.
Limitations of our study are: 1) some family histories are missing. When medical records did not specify onset of cancer age and relationship from patient, telephone interviews were done. But because of memory loss and reluctance to reveal family secrets, they were not complete; 2) IHC is just screening test. Additional MSI, methylation and gene sequencing tests will confirm germline mutations. Although we must consider the high methylation rate of MLH1 and the sensitivity of staining, we propose a high probability of Lynch syndrome in patients with both sites negative for MLH1 (4/8; cases 1-4 in Table 4).
In summary, 9% (3/32) of women with synchronous gynecologic tumors had either clinical or IHC criteria suggestive of Lynch syndrome. Therefore, we conclude that identifying suspicious Lynch syndrome patients though acquisition of family history and IHC can prepare us for better consultation and efficient prevention of multiple cancers.




 XML Download
XML Download