

 PDF
PDF ePub
ePub Citation
Citation Print
Print


I. Introduction
Next-generation sequencing (NGS) technology now provides a cost-effective approach to the large-scale sequencing of human samples for medical and population genetics. Within the field of personalized genomics, ambitious sequencing projects such as the 1000 Genomes Project [1], the Cancer Genome Atlas (http://cancergenome.nih.gov/), and the International Cancer Genome Consortium (http://www.icgc.org/) are seeking to identify genomic variants among human genomes and to use this knowledge to determine the genetic underpinnings of human diseases by associating variants with symptoms. Several bioinformatic methods handling NGS data, including sequencing reads alignment, variant detection, and visualization of read sequences, have been developed. However, when considering that an important goal of NGS data is deciphering human genome sequences, it is imperative to annotate detected variants in terms of disease-related phenotypes.
Recently, tools designed for annotating variants in NGS data have been introduced, such as ANNOVAR [2], SeqAnt [3], and GAMES [4]. These tools annotate variants specifically with respect to genes harbouring variants, functional importance scores, and evolutionary conservation. ANNOVAR is a command-line tool that uses information from the UCSC Genome Browser to provide annotations. The SeqAnt is Web-based and can be downloaded, and it also relies on resources from the UCSC Genome Browser. The GAMES supports exome-Seq mutation discovery and functional annotation using the UCSC Genome Browser. All of them can place single nucleotide polymorphisms (SNPs) into functional classes, describe nearby genes, and indicate which SNPs are already described in dbSNP. None of them provides disease-related phenotype information, gives expression probe information for downstream analysis using microarray, or supports gene (or disease)-centric queries (Table 1).
In this article, we focus on disease-related phenotype annotations necessary for the analysis of variants. It is essential to develop a computational environment that can assess the likely functions of the variants observed, and whether or not they are present in existing variant databases. AnsNGS (annotation system of sequence variations for next generation sequencing data) is a new tool for annotating detected variants using disease-associated variants.
II. Methods
1. Input Files of the AnsNGS
Sequencing data can be uploaded and the results can be viewed using an interactive, Web-based graphical user interface (GUI) that has been tested for standard browsers. The AnsNGS takes text-based input files, where each line corresponds to one genetic variant. In each line, the first five tabdelimited columns represent chromosome, start position, end position, the reference nucleotide, and observed nucleotide. Additional columns can be supplied and will be printed in identical form in output files.
2. Genome-Wide Variant Annotation
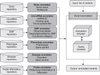
The AnsNGS is a Web-based system that can comprehensively annotate variants detected with NGS data to provide a multifaceted approach for disease-related phenotype. The AnsNGS queries various tables in the UCSC Genome Browser [5], which provides a MySQL database annotation or any data set conforming to generic feature format ver. 3 (GFF3). As shown in Figure 1, the system consists of six annotators. The six annotators in the AnsNGS are a gene annotator, a microRNA annotator, an SNP annotator, a phenotype annotator, a disease annotator, and a probe annotator.
The gene annotator uses the refFlat table in the UCSC Genome Browser to obtain the genomic location of the nucleotide, the chromosome, the encoded protein(s), the gene, and the exon count (for all available isoforms). We use this information to determine the position of the mismatch in the gene and to determine whether it is in a coding/noncoding, exon, intron, untranslated region (UTR), or intron/exon junction region (intronic regions contiguous to exon starts and exon ends that are important to evaluate splicesite mutations).
The microRNA annotator employs the miRBase [6] to report microRNAs harbouring variants. The miRBase released in April 2010 includes 940 human microRNAs. From miRBase, the AnsNGS extracts microRNA names, host transcript genes, and genomic locations (e.g., intron, exon, intergenic, 5' UTR and 3' UTR).
The SNP annotator uses the snp130 table in the UCSC Genome Browser. The snp130 table contains almost 19 million SNP annotations, and it includes the first set of SNP calls from the 1000 Genomes Project. The tables are structured to include the position of the SNP on the genome assembly as well as additional information, such as sequences of the observed alleles from rs-fasta files, genotype counts, allele frequencies, kinds of mutation, and the pathogenetic significance of the SNPs, if reported.
The phenotype annotator uses the Genetic Association Database (GAD) which collates known polymorphism-disease associations [7] to provide disease-related phenotype information. Polymorphism-disease association data curated in this way are likely to comprise markers that occur in linkage disequilibrium with the presumed disease associated/functional variants.
The disease annotator takes omimGeneMap and omimMorbidMap tables in the UCSC Genome Browser. The AnsNGS provides annotations of known disease genes and the disease relevance of candidate genes using OMIM [8]. This type of information is critical for the proper analysis of whole-exome sequencing projects, in which mutations in different genes might be biologically involved in the same disease.
To connect NGS data analysis and microarray data analysis for the post hoc approach, reliable functional annotation of microarray probes is essential for the analysis and interpretation of the biological processes. The probe annotator provides annotation linking oligonucleotide probes to target genes, and it is essential for functional biological analysis of Affymetrix microarray experiments.
3. Annotated Output Files
Annotated output data can be displayed in several ways. The GUI provides a simple and convenient interface for the user to select the annotation information associated with a given variant or to highlight the variants by functional classes. The user is also able to download the annotated variation in a tab delimited text file format. The specific annotation field outputs are found in Table 2.
4. Validation Method
A total of two sequence datasets of various sizes and types were annotated by the AnsNGS. The first consisted of a 48-kb region, including the fragile X mental retardation 1 (FMR1) locus [9]. The second consisted of four HapMap exomes for Freeman-Sheldon syndrome [10]. Sequence data were uploaded to the AnsNGS website. The AnsNGS program determined the annotation information for the variable sites. The resulting output can be viewed on a Web-based GUI that is tested for standard browsers, in a series of downloadable tab-delimited text files (Figure 2).
III. Results
As a direct demonstration of the utility of the AnsNGS for detecting functional variants, we annotated the data from a 48-kb resequencing experiment of a single human sample with a known coding sequence mutation at the FMR1 locus [9]. A total of 37 variant sites were identified and annotated in 0.13 seconds (based on searches with a single Intel(R) Xeon(R) CPU, 3.00 GHz), including the I304N mutation, which has been shown to result in intellectual disability. The AnsNGS showed that the variants are associated with mental retardation, X-linked, FRAXE type (OMIM ID: 309548). Disease-related phenotypes were global DNA methylation defect as well as ataxia and cognitive function. In terms of pathophysiology, a loss or shortage of FMRP disrupts the normal functions of nerve cells and, consequently, the nervous system, causing severe learning problems, intellectual disability, and the other features of fragile X syndrome. An FMR1 gene mutation and the characteristic signs of fragile X syndrome also contribute features of autism spectrum disorders that affect communication and social interaction. In Human Genome U133A 2.0 Array (Affymetrix, Santa Clara, CA, USA), genes harbouring variants correspond to Affymetrix probe ID 203689_s_at and 215245_x_at.
To illustrate the utility of the AnsNGS in identifying causal genes for Mendelian diseases with dominant inheritance, we used whole-exome data sets. We downloaded the Freeman-Sheldon exome data for eight HapMap subjects. We then extracted the exome data for the first four subjects, including two Yoruba subjects (NA18507 and NA18517) and two European-Americans (NA12156 and NA12878). The AnsNGS showed that the variants are associated with arthrogryposis (OMIM ID: 160720). Disease-related phenotypes were carcinoma, squamous cell, and esophageal neoplasms in the Medical Subject Heading (MeSH) disease term. In Human Genome U133A 2.0 Array (Affymetrix), genes harbouring variants correspond to Affymetrix probe ID 205940_at. These results demonstrate the utility of annotating genetic variations with terms of disease-related phenotypes.
We have developed and validated the AnsNGS, a novel application for the annotation of NGS data with respect to disease-related phenotypes. Considering that it is important to identify induced variants responsible for a mutant phenotype directly, the AnsNGS is useful to discriminate silent mutations or polymorphisms from variants that are potentially associated with a phenotype or a disease. The AnsNGS generates concise and highly readable reports for a functional selection of important genetic events. In fact, the AnsNGS takes text-based input files, where each line corresponds to one genetic variant, including SNPs, insertions, deletions, or block substitutions.
IV. Discussion
Understanding the underlying complexity of disease phenotypes is a key to identifying causative mutations from NGS data. The annotation data reported by the AnsNGS can meet this requirement. Integrating the AnsNGS into a human DNA sequencing pipeline is powerful and versatile in clinical applications of NGS. The AnsNGS overcomes a significant bottleneck that can slow the wide-scale application of NGS for a host of genetics research and clinical genetics applications. The AnsNGS allows researchers to significantly narrow down the genomic regions of interest to their research, making an efficient and time-saving solution for everyone working in the NGS area and a perfect fit for any NGS analysis pipeline.
In conclusion, the AnsNGS is not only a new application for mining functional SNPs, insertions, and deletions from NGS data, but it also aids in the overall interpretation of large-scale sequencing data. The AnsNGS supports the identification of disease-associated genes and the prioritization of genes relevant to other diseases. The ultimate purpose of the AnsNGS is to provide biological insight into genetic events of disease-linked variants. The AnsNGS can lead to a translational bioinformatics approach and a paradigm shift in the aspect of biological interpretation, particularly in terms of the omics mechanisms of personalized medicine.



 XML Download
XML Download