

 PDF
PDF ePub
ePub Citation
Citation Print
Print


I. Introduction
Pulmonary studies have reported that diffuse parenchymal lung diseases (DPLD) account for around 130-180 disorders of the lung tissue affecting the lung parenchyma, often with very nonspecific symptoms [1,2]. Demedts et al. [3] reported that the prevalence of idiopathic pulmonary fibrosis (IPF) ranges from 6 to about 14 per 100,000 person-years, and the incidence of IPF reaches to 175 cases per 100,000 person-years in those aged 70 years or older. Thus, its importance is increasing in Korea as the society ages.
Interstitial lung diseases (ILD) refer to a group of diseases that invade the interstitial tissues of the lung, excluding malignant tumors and infections. ILDs invade not only the interstitial but also noninterstitial tissues, such as the trachea and pulmonary artery, and therefore are often called "diffuse parenchymal lung diseases (DPLD)" or "infiltrative lung diseases (ILD)." DPLD is increasingly used as a generic term for these disorders, in preference to terms such as interstitial lung disease or diffuse lung disease [4]. The joint statement of the American Thoracic Society (ATS) and the European Respiratory Society (ERS) defines IPF, the most common of several idiopathic interstitial pneumonias (IIPs) in DPLDs, as a specific form of chronic fibrosing interstitial pneumonia, limited to the lung, that is progressive and usually fatal and unresponsive to treatment [5]. It is a specific entity with the histopathological pattern of usual interstitial pneumonia (UIP) on surgical lung biopsy [6] and a median survival of 3 years [7]. Clinical IPF indicates relatively undesirable catamnesis and is recognized as a distinct clinical disorder from other DPLDs.
Within the IIPs, it is important to distinguish between IPF and the other interstitial pneumonias. The importance of a detailed differentiation of IPF from other IIPs has been demonstrated by Drent et al. [8]. IPF can be distinguished from other forms of DPLD by clinical presentation, lung function test, high-resolution computed tomography (HRCT) findings, chest radiology features, laboratory studies, and pathology. Precise and correct diagnosis for the degree of inflammation and fibrosis of IPF is important for prognosis and treatment, but many of the diseases are rare, and establishing a differential diagnosis for ILD is considered difficult [2]. Due to the difficulties in the differential diagnosis in terms of clinical approach, diagnostic uncertainty, and philosophy between IPF and nonspecific interstitial pneumonia (NSIP), previous clinical reports on IPF must be considered carefully. It is difficult even for respiratory specialists to decide to what extent examinations should be undertaken.
However, physicians only performing physical examinations often fail to make a correct diagnosis and prescribe curative methods for DPLDs, as it is hard distinguish these from other lung diseases, such as pulmonary tuberculosis or chronic obstructive pulmonary disease (COPD), including chronic bronchial infection, emphysema, and symptoms by aging. The American Thoracic Society/European Respiratory Society (ATS/ERS) consensus statement [9] described the major and minor criteria for the clinical diagnosis of IPF as developed from expert opinion in the absence of a surgical lung biopsy. It includes HRCT and bronchoaveolar lavage (BAL) among the 4 major criteria, and increases the likelihood of a correct clinical diagnosis of IPF [10]. If patients do not meet these major and minor criteria, the respiratory physician should be directed to perform a surgical lung biopsy [3,5,11]. Table 1 shows the major and minor criteria for the clinical diagnosis of IPF from the ATS/ERS consensus statement.
Physical examination of a patient in DPLDs is frequently abnormal, but with nonspecific and differential findings. In Korea, the prevalence rate of DPLDs is deemed to be high, although no accurate values have been reported because of the difficulties in differential diagnosis among the DPLD groups in primary care.
We performed knowledge modeling for the diagnosis of IPFs through knowledge working groups composed of respiratory specialists and radiologists. For knowledge modeling in this study, diagnosis knowledge acquisition, refining, and representing were performed iteratively. The knowledge was acquired from all resources, including pulmonary disease guidelines and physicians' experience. For the knowledge refining process, acquired knowledge was applied to clinical cases and analyzed by the chi-square test. The refined knowledge was represented with IF-THEN rules for the inference engine of the integrated clinical decision support systems (CDSSs) developed in this study.
Meanwhile, integrating a medical database with an expert system improves the knowledge base construction and CDSS performance. Yet, the IT environment is still heterogeneous and most of the diagnostic systems have been developed as stand-alone systems. This has been considered a major impediment to knowledge acquisition for CDSSs. Knowledge acquisition directly from the database through the interface mechanism is often of considerable value to CDSS quality management [12]. The efficiency and quality of the CDSSs could increase when medical instruments, such as the laboratory interpretation system, electrocardiogram, electroencephalogram, magnetic resonance imaging, and HRCT, are integrated with them. Medical instruments are indispensable to the practice of medicine and essential to correct diagnosis. Integrated medical instruments that have internal computers containing the CDSSs are not difficult to use with current interfacing technology.
In this study, the IPF CDSS linked with the Picture Archiving Communication System (PACS) automatically acquires the interpretation and radiological diagnosis results of HRCT through the computer vision system (CVS) and DPLD CDSS. The DPLD CDSS integrated with the CVS was developed and evaluated in our previous study [13]. The CVS automatically interprets ground-glass opacities (GGOs) and honeycombing, some of the main HRCT image findings required to diagnose IPF. The DPLD CDSS deduces the information necessary for diagnosing 12 DPLD-related diseases, using the image findings automatically interpreted by the CVS. The CVS calls the HRCT images, containing the image files transmitted from the PACS, from the directory and begins image detection of the lesions of the GGOs and honeycombing. A digital imaging and communications in medicine (DICOM ) [1] data file is composed of names, gender, and various associated patient data necessary to the CDSSs. In reading such DICOM files, the information is put into the knowledge base used in the CDSS. Data warehousing has been proved as being an efficient and effective information supply from heterogeneous systems in much previous research and many technical reviews [14,15]. We attempted to integrate all of the patient information in the hospital information systems by constructing a data warehouse.
The purpose of this study is to acquire knowledge from HRCT images for diagnosing DPLD and knowledge from knowledge working groups for diagnosing IPF, and to develop an IPF CDSS integrated with other hospital information systems and DPLD CDSSs [16] to make accurate differential diagnoses between IPF and NSIP. In addition, the study was designed to enhance feasibility by applying systems to real clinical settings and physician evaluations.
II. Methods
1. Collecting Clinical Cases
In this study, the clinical and radiological screening diagnosis of IPF was subjected to system development. Two hundred ninety cases were diagnosed as DPLDs or IPFs and treated from December 2002 to February 2008 at Seoul National University Hospital and Yonsei University Hospital. One hundred nine of these patients (Seoul National University Hospital, 7; Yonsei University Hospital, 102) who sufficiently underwent diagnostic examination were included in the study. Among the patients whose diseases were not definitely diagnosed histologically, those having heart or renal failure that might influence radiological or clinical findings and those having other pulmonary diseases (lung cancer, chronic obstructive pulmonary disease) were excluded from this study.
2. Diagnostic Knowledge Acquisition from Working Groups
Expert knowledge on IPF diagnosis was acquired by conducting interviews with respiratory specialists, radiologists, medical domain engineers, and system developers at regular group meetings. Knowledge modeling began with collecting knowledge from various pulmonary disease guidelines. The knowledge working group collected and defined diagnostic knowledge for DPLD and IPF, based on clinical experiences and medical records, through periodic study meetings. Simultaneously, websites, journals, and textbooks were studied with the intent to acquire up-to-date data and knowledge.
To refine the collected knowledge, we added and verified respiratory physicians' experience and clinical practice knowledge and applied it to the IPF CDSS diagnosis rules. These rules were applied to confirmed cases for validation. The differences shown in the respective disease groups were statistically analyzed by the chi-square test, and the information that showed a statistical significance was given clinical significance on the basis of respiratory specialist experience and knowledge. Rules recognized for providing clinically meaningful differences, which had been verified for validity based on clinically confirmed cases, were aligned with the actual diagnosis process and expressed in IF-THEN form for each stage of the process, according to the information acquisition time and method and importance of the differentiation priority.
Since the defined knowledge was designed to focus on increasing discrimination power, it was built by including as many patients as possible with the disease in the initial stage of the system, and thereafter excluding cases showing a clear decrease in possibility. The reasoning method consisted of repeatedly narrowing the range of target diseases with each condition added to the entire range of diseases, until the final diagnosis was reached.
In this study, however, we did not include surgical lung biopsy results in the diagnosis process of the IPF CDSS. If a final decision could not be made using other major and minor criteria testing results, the IPF CDSS would recommend a surgical lung biopsy as the next test procedure and physicians would carefully make a decision based on the patient's state.
The knowledge base was reconstructed by rearranging the knowledge collected and arranged in that base. The final diagnosis was made after information for diagnosing DPLD and IPF was acquired through the patient's medical history, pulmonary examination, radiological examination, BAL, and lung biopsy. As it was difficult to extract from the examination reports, the "insidious onset of otherwise unexplained dyspnea on exertion" was excluded from the criteria. A comprehensive knowledge base was rebuilt, which included the clinical and image findings required for the IPF diagnosis standard as presented by the joint statement of the ATS/ERS [3,11] and IPF diagnosis presented by Hunninghake et al. [17].
3. Integrating CDSSs with PACS
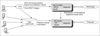
The data warehouse we constructed functions as an integrated hospital information system that potentializes the final clinical diagnosis and HRCT diagnosis. Figure 1 presents the integration architecture of 2 CDSSs and other hospital information systems, including PACS, using the CVS and data warehouse. The data warehouse in Figure 1 consists of HRCT image data from PACS, patient clinical information, patient physical examination results, and laboratory test results. The data mart could be handled with multidimensional online analytical processing (OLAP), being linked with the database of HRCT data and knowledge base of the CDSSs for DPLD and IPF diagnosis. It processes various and complicated queries requested by CDSSs. We used Microsoft SQL Server 2000 to develop the data warehouse and Microsoft Analysis Services to analyze the clinical data from the order communication system (OCS), laboratory information system (LIS), and other hospital information systems.
The DPLD CDSS integrated with the CVS was developed in our previous project and deduced diagnosis from 12 DPLD disease classifications [13]. The IPF CDSS helps respiratory specialists to diagnose IPF by using collected patient information from the data warehouse integrating hospital information systems. The interface and inference engine for the IPF CDSS was developed using .NET with the Visual Web Developer 2005 express edition and extensible markup language (XML), web standard language.
We developed an optional function to integrate the IPF CDSS with the CVS. In clinical practice, a respiratory physician makes a diagnosis with the help of a radiologistp the CS, using the a comprehensive knowledge base was rebuilt, which included tth automatic HRCT interpretation results from the CVS. The respiratory physician can access the information from the CVS.
III. Results
1. Collecting and Defining Diagnosis Knowledge for IPF
Expert knowledge regarding IPF diagnosis was acquired by holding interviews with respiratory specialists, radiologists, medical domain engineers, and system developers at regular group meetings.
IPF, also termed cryptogenic fibrosing aveolitis (CFA) in Europe [18], is a distinct and specific form of chronic fibrosing interstitial pneumonia with no known cause. The ATS/ERS revision of the classification of DPLD [11] established the guidelines for the classification of IIPs, distinguishing between IPF and other IIPs [1]. Since they have a clinical similarity but their clinical courses and responses to therapy are different, the diagnostic process in patients with suspected DPLD begins by making the distinction between idiopathic and nonidiopathic diseases. For example, the diagnosis of sarcoidosis needs a compatible clinical picture of a systemic disease, histological demonstration of noncaseating granulomas, and exclusion of other diseases capable of producing a similar histological or clinical picture [19].
The common clinical feature of IPF is insidious progressive shortness of breath or dyspnea for at least 3-4 months [5,11]. Cough and minimal or no sputum is a general feature of IIPs. The presence of clubbing on physical examination is helpful in suggesting a diagnosis of IPF and pulmonary fibrosis associated with rheumatoid arthritis, asbestosis, or fibrosing NSIP [18]. A thorough clinical diagnostic approach begins with a detailed medical history, physical exam, PFTs, chest x-ray, lung function studies, blood tests, and tissue analysis [5,11]. An important goal of these multidisciplinary clinical approaches is the early diagnosis of the disease, which is enhanced by diagnostic agreement [16,20].
If clinical findings fail to suggest an alternative diagnosis, HRCT image findings should be obtained [21]. HRCT scanning has greatly improved the ability to visualize and characterize abnormalities in IPF. Extensive reticulation, traction bronchiectasis, honeycombing cysts, a subpleural predominance, and a somewhat patchy distribution are strongly suggestive of HRCT findings in IPF. The appearance of GGOs on HRCT is not characteristic of IPF [5]. Therefore, HRCT allows the clinician to make the distinction between "possible IPF" and "non-IPF" [5,18]. Hunninghake et al. have suggested that relatively accurate clinical and radiologic data, interpreted by an experienced pulmonologist or radiologist, are sufficient to diagnose IPF, precluding and even obviating the need for a lung biopsy [10,18].
Transbronchial biopsy (TBBx) and BAL have limited use as diagnostic tools, but are sufficient to help exclude other causes of pulmonary interstitial infiltrates, such as sarcoidosis, hypersensitivity pneumonitis, malignant disease, and infection, when surgical biopsy is not an option [5]. A recent article in Chest [19] suggested that the TBBx may have some utility with well-defined criteria. Berbescu et al. [21] examined TBBx samples from 22 UIP patients.
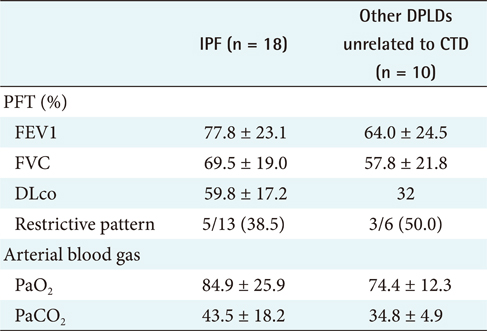
Restrictive pulmonary physiology is the classic finding on pulmonary function testing in IPF [20,22]. There is impaired gas transfer reflected in a decrease in carbon monoxide diffusion in the lung (DLCO) and alveolar oxygen partial pressure (PaO2), which is associated with increased mortality [18,23]. An increase in oxygen desaturation on exercise is well correlated with the extent of disease on HRCT. There may be hypoxemia with an increased alveolar-arterial O2 gradient (A-aPO2). In addition, although the typical findings of pulmonary function tests (PFTs) in IPF are consistent with restrictive impairment, PFTs or arterial blood gases (ABGs) may be normal. Typical findings of airway restriction include a decreased forced vital capacity (FVC) and total lung capacity (TLC) and DLco [3,5,11].
The presence of typical findings on clinical and radiologic examination may allow for a confident diagnosis, precluding the need for a surgical lung biopsy. A surgical lung biopsy in suspected IPF remains the gold standard for diagnosis, but it can be limited to those patients who show an atypical pattern, such as a predominant lymphocytosis on HRCT or in BAL, or an atypical clinical presentation, such as young age or short duration of illness. The surgical lung biopsy should be carefully considered and performed on the bases of HRCT findings [5].
In this study, we did not include surgical lung biopsy results in the IPF CDSS diagnostic process since the diagnosis of the CDSS focused on differentiating IPF from other DPLDs in primary care. Instead, the IPF CDSS recommends surgical lung biopsy as the next test procedure, and that physicians make a careful decision according to the patient's state.
2. Refining Diagnosis Knowledge for IPF
For knowledge redefinition, we added verified respiratory physicians' experience and clinical practice knowledge to the IPF CDSS diagnosis rules. The collected knowledge was applied to cases of confirmed diagnosis for validation. The information that showed a statistical significance was given clinical significance on the basis of the respiratory specialists' experience and knowledge.
The collected knowledge based on major and minor criteria for the clinical diagnosis of IPF suggested by the joint statement of the ATS/ERS guidelines [3,11] and HRCT image findings for IPF diagnosis suggested by Hunninghake et al. [17,24] was refined and redefined. The differences shown in each DPLD group were statistically analyzed by the chi-square test, which was applied to differentiate IPF from other DPLD groups unrelated to connective tissue disease (CTD). In this analysis, we excluded an unexplained insidious dyspnea because it is currently difficult to acquire such cases.
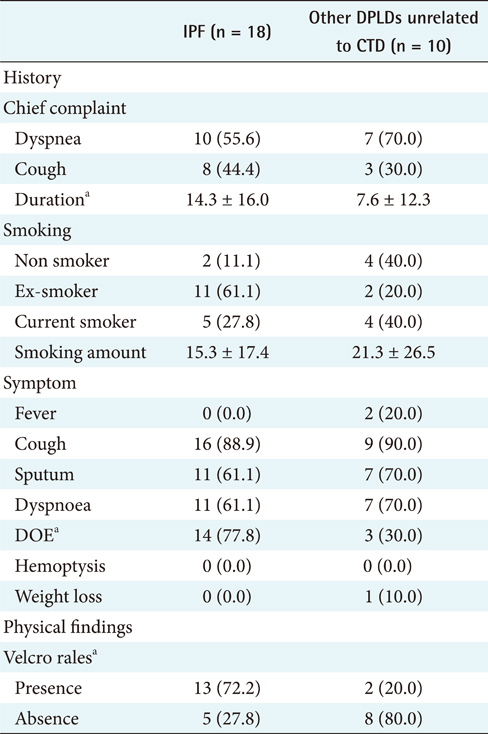
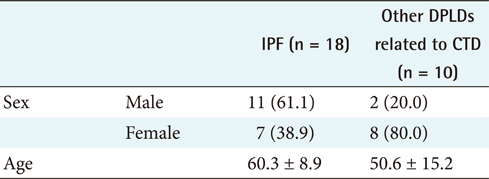
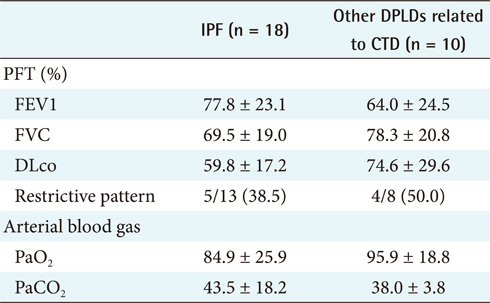
For the first step, to differentiate IPF from CTD unrelated to other DPLD groups [2], the 18 IPF cases confirmed by diagnosis through surgical biopsy were compared with the 10 cases of other DPLD groups confirmed clinically (Table 2). The characteristics of these 2 diseases are apparently different in a population.
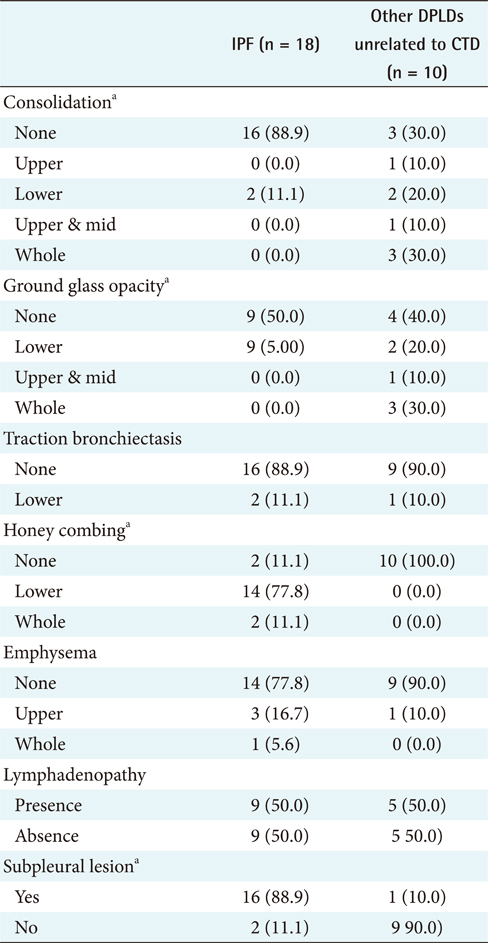
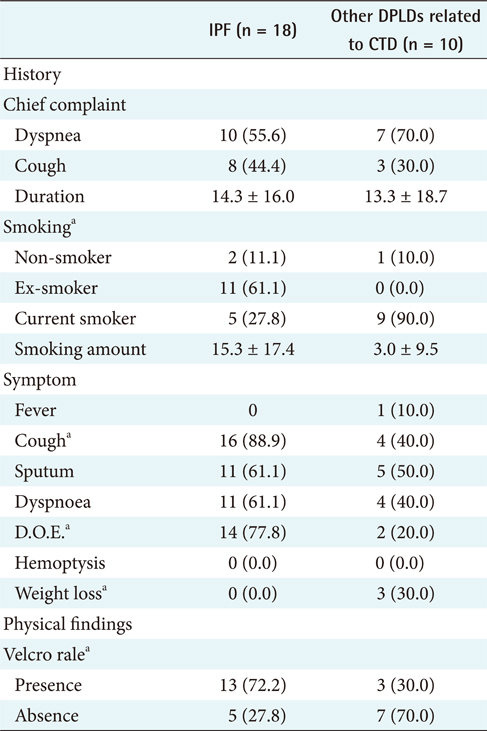
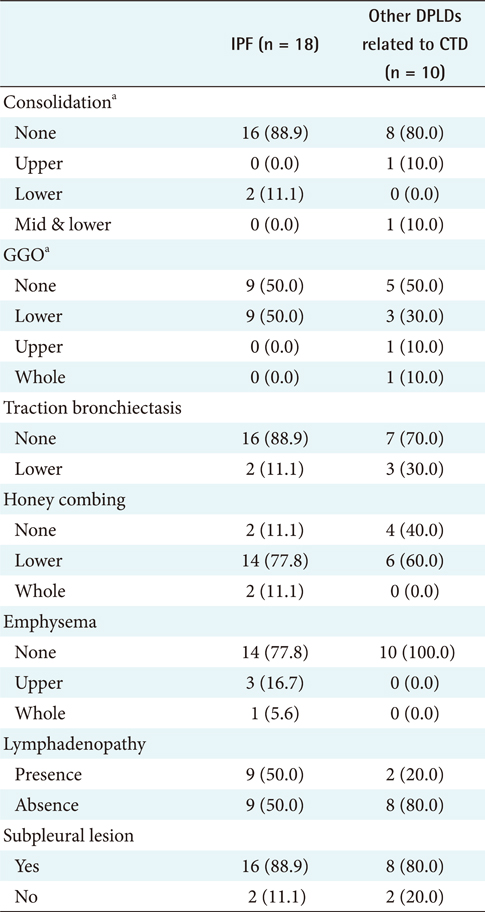
The different clinical findings between IPF and other DPLD disease groups were analyzed by the chi-square test. A p-value lower than 0.05 was considered statistically significant. The results (Appendixes 1 and 2) show that, among the clinical features, dyspnea on exertion (DOE) (IPF, 77.8%; other DPLDs unrelated to CTD, 30.3%; p < 0.05), duration from the beginning of having symptoms to visiting clinics (IPF, more than 14 days; other DPLDs unrelated to CTD, less than 12 days), and the presence of Velcro rale (IPF, 72.2%; other DPLDs unrelated to CTD, 20.2%; p < 0.05) were apparently different between IPF diseases and other DPLDs unrelated to CTD. The radiologic findings analyzed by the chi-square test demonstrate that consolidation, traction bronchiectasis, GGO, lower location of honeycombing, and subpleural lesions were significantly different between IPF and other DPLDs unrelated to CTD (p < 0.05) (Appendix 3).
To differentiate IPF from other DPLDs related to CTD, however, the 18 IPF cases confirmed with the diagnosis of surgical biopsy were compared to 10 cases of other DPLDs related to CTD (Appendixes 4 and 5). The characteristics of these 2 diseases have similarities in radiological diagnosis, but are apparently distinguished by clinical features and prognosis by a secondary cause of DPLD. The different clinical findings between IPF and other DPLDs related to CTD were analyzed by the chi-square test. A p-value lower than 0.05 was considered statistically significant. The different clinical findings between IPF and other DPLDs related to CTD were analyzed by the chi-square test. The results (Appendix 6) show that among the clinical features, ex-smoker (IPF, 11.1%; DPLDs related to CTD, 0.0%; p < 0.05), cough (IPF, 88.9%; DPLDs related to CTD, 10.0%; p < 0.05), DOE (IPF, 77.8%; DPLDs related to CTD, 40.0%; p < 0.05), weight loss (IPF, 11.1%; DPLDs related to CTD, 0.0%; p < 0.05), and the presence of Velcro rales (IPF, 72.2%; DPLDs related to CTD, 30.0%; p < 0.05) were apparently different between IPF diseases and other DPLDs related to CTD. The radiologic findings analyzed by the chi-square test (p < 0.05) show that consolidation, traction bronchiectasis, GGO, lower location of honeycombing, and subpleural lesions were significantly different between IPF and other DPLDs related to CTD (Appendix 7).
When the collected knowledge from guidelines such as the joint statement of the ATS/ERS and refined knowledge by statistical analysis were applied to 18 cases confirmed as IPF, the former diagnosed 1 case (6%) and the latter diagnosed 14 cases (78%). If traction bronchiectasis, one of the HRCT image findings, was removed from the diagnosis rules, the diagnosis performance of the collected knowledge was enhanced by 6 cases (33%). In addition, when the collected knowledge from guidelines such as the joint statement of the ATS/ERS and refined knowledge by statistical analysis were applied to 11 cases under medical treatment, the former diagnosed 1 case (9%) and the latter diagnosed 7 cases (64%). If traction bronchiectasis, one of the HRCT image findings, was removed from the diagnosis rules, the diagnosis performance of the collected knowledge was enhanced by 3 cases (27%). Therefore, refined knowledge performed better than collected knowledge.
3. Representing Diagnosis Knowledge for IPF
The 3 diagnosis steps for IPF were extracted from refined knowledge. Figure 2 shows the flow chart of these steps, which we refined from the respiratory physicians' experience and clinical practice knowledge.
As the order of priority of the 3 steps was not provided in IPF diagnosis research literature, the order of priority in the diagnosis process was designed based on the empirical knowledge of experts verified by clinical trials at the redefinition stage of this research, and was reflected in the diagnosis process of CDSS.
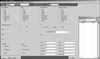
Step 1 is the step for confirming the presence of clinical features. When the program is run by calling a patient's clinical information from the data warehouse through the data interface forms or with the manual input of information, and if, based on that basic clinical information, there is a chance of IPF, the clinician proceeds to the next step. If the likelihood of IPF is small, clinical information enhancement or PFT implementation is recommended. After the PFT result is input into the system, the next process is suggested based on the result. In step 2, the step diagnosing radiological features, the HRCT image findings and reading results are automatically received as direct input from the DPLD CDSS and CVS. From the DPLD CDSS, the diagnosis results confirmed by the radiologist are received as text. For high-level users able to read the information, in addition to the text reading option, direct image examination is made available as a diagnostic reference. When the clinical information and radiological information are ready, the likelihood of IPF is analyzed and the physician either modifies the information or proceeds to the next step. Step 3, the step diagnosing the presence of CTD-related DPLDs, after selecting the target group with a high likelihood of IPF, confirms the CTD-related DPLDs in order to eliminate the possibility of other associated diseases. Verification of antinuclear antibodies (ANAs) and rheumatoid factors, the markers for CTDs, is strictly enforced, and proceeding to the next step is allowed only after the physician confirms whether there are any associated CTDs present. Step 4, the diagnosis step to eliminate the presence of other associated diseases, as in cases of CTDs, eliminates cases of important associated lung diseases that are expected to bring substantial change to the prognosis and treatment guidelines. If lymphocytosis (in terms of the BAL differential count, lymphocyte > 15%) is shown from BAL, even if it clinically falls under IPF, the user is shown other diseases so that they can also be considered. Since the likelihood of IPF decreases for people under the age of 50 years, the user is given the opportunity to consider other associated diseases. The likelihood of IPF as the final inferred diagnosis result is presented for the data, which has been confirmed by the user in a conversation form. For the CDSS inference application, the knowledge is expressed with IF-THEN rules.
4. Developing IPF CDSS and Integrating with DPLD CDSS
Patient clinical information from the OCS, past diagnostic information, physical and laboratory test information, HRCT image information, and knowledge and experience based on inquiry are needed in order for respiratory specialists to clinically diagnose IPF. Thus, hospital information systems should be integrated so that such diagnostic processes, as well as the process of knowledge acquisition, can be implemented in the system.
The diagnosis process of the IPF CDSS developed in this study is as follows (Figure 3). When the IPF CDSS program is started, a patient case list, which already exists in the data warehouse, appears on the CDSS. On clicking a patient name or number in the list, all laboratory examinations from the data warehouse, including clinical information and the diagnosis results from the DPLD CDSS of the given patient, are read into the IPF CDSS and displayed on the screen. The input of the confirmed diagnosis result among the 12 diseases from the DPLD CDSS, based on the image interpretation from the CVS, are both automatically and directly made.
If an examination required for IPF diagnosis is missing, the program recommends the given examination, carries out the diagnosis when all the information needed for that diagnosis has been input, and presents the diagnosis confirmation for the physician to review. If making the final diagnosis is difficult based on the given result, the system gives the physician the option to recommend surgical lung biopsy, based on the patient's condition.
5. Validation and Evaluation of CDSSs
IPF CDSS validation was implemented. First, the IPF diagnosis algorithm validation was implemented for the filtering process to express the diagnosis knowledge as a rule.
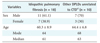
To verify the validity of the IPF diagnosis algorithm, 290 histological confirmation cases of DPLDs were collected for validation of the diagnosis rules and algorithms for 3 years (2005. 2.-2008. 2.) from Seoul National University Hospital and Severance Hospital in Yonsei University. The cases included HRCT image data, clinical laboratory test results, patient physical test results, and lung biopsy results. Out of the 109 cases that were critically diagnosed as IPF and being treated, 81 (74.3%) were diagnosed as IPF by the IPF CDSS. Likewise, the IPF CDSS was applied to the 28 cases that were diagnosed as other DPLDs. As a result, there were no cases where other diseases had been diagnosed as IPF (Table 3).
Meanwhile, validity analysis of the CVS-integrated CDSS was implemented with 10 patient cases diagnosed as IPF. The system detected 100% honeycombing from the HRCT images of the 10 patient cases, and the CDSS using the IPF diagnosis rules showed an accuracy rate of 98%. Considering such results, the IPF CDSS developed in this study, with its accurate IPF diagnosis decision-making, is considered able to aid in the quality of diagnosis.
For the evaluation of the CDSS, the DPLD CDSS was set in a clinical environment. Five radiologists at Seoul National University Hospital and 2 respiratory physicians at Yonsei University Hospital were educated to use the CDSS program, and evaluated the system usability. The evaluation of demonstrative operation was analyzed. When ascertaining the precision of diagnosis, the automatic image detection of the CVS and radiologists' gross examination indicated a consentaneity of about 99%.
A questionnaire was conducted on user satisfaction, qualitative improvement, and proposals, among other things, to examine the outcomes after clinical experiment. The users mostly felt that the CDSS would be of help to diagnose diseases and most of them were satisfied with the results. Many users suggested that HRCT findings should be classified as to meaning rather than presented in alphabetical order. Surveys and system use evaluations will be regularly collected and reflected iteratively for system modification, enhancement, and improvement to increase the accuracy and efficiency of diagnosis.
IV. Discussion
The significance of this study is in developing the CDSS by acquiring and redefining the knowledge needed for IPF diagnosis. This study undertook knowledge modeling of the knowledge needed for IPF diagnosis, based on study of the literature, respiratory specialists' experience, and analysis of confirmed cases, stored the defined knowledge in a knowledge base, and expressed the knowledge with IF-THEN rules. To derive the new diagnostic rules for IPF diagnosis, knowledge modeling was done through a process consisting of knowledge collection, knowledge refining and redefining, and knowledge representation. First, the knowledge working group, through a study of the literature, compiled the knowledge for understanding the individual data sets needed for differential diagnosis, such as the location and duration of the disease, symptoms and indications, and various examination results. For knowledge refining and redefining, the differences between each data set's disease groups were statistically analyzed through a chi-square test, and for each data set showing statistically meaningful differences, clinical significance was assigned based on the experience and knowledge of a respiratory specialist.
The CVS-integrated DPLD CDSS was integrated with the IPF CDSS developed in this study, and the automatic analysis results of the image data were automatically received as direct input. The HRCT image of PACS directly received as input from the CVS were GGO and honeycombing opinions. The algorithm allowed the next step when 2 of the 4 criteria were satisfied. It was also significant in terms of integrating the CDSSs, which were verified and evaluated by the clinical demonstration.
If forecasting power can be improved through knowledge base supplementation, it is hoped that, for limited situations, this will develop into a basis for treatment. In such cases, the latest knowledge and experience related to the treatment and control of relevant diseases must be offered together in some way. This advancement requires the appropriate management of a histologically confirmed case database, and since this is needed to derive more elaborate diagnosis rules in the future, it is recommended that the recognition of new data be regularly confirmed by respiratory specialists and this process be managed separately.
In this study, new images were received during the demonstration period and used to validate the prospective results of the system. By validating the usability and convenience of the system, this study demonstrates that it can be set up and expanded from the demonstration run for full implementation. By directly applying the developed system in a clinical environment, it was possible to measure its actual utility in terms of accuracy, convenience, and usability for diagnosis decision-making of radiologists and respiratory specialists. This paper is part of a larger ongoing study. The demonstration run period will be extended to iteratively and regularly check the accuracy of diagnosis and demands of end-users, and the results will be reflected in system improvements.



 XML Download
XML Download