

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Alzheimer's disease is one of the most serious medical problems of the modern era, given the rapidly aging global population. This disorder may accompany various neurological deficits, including failure of memory formation, loss of cognitive function, and change in personality. Senile plaques, neurofibrillary tangles, and a wide range of neuronal deaths are well-known common pathological findings in lesions associated with this disease.1 β-amyloid peptide (Aβ), a main component of senile plaques, is known to be the most important causative factor.234 Therefore, most studies have focused on the formation, deposition, and neurotoxic effects of Aβ, including exploring their prevention and delay.5 Oxidative stresses have been implicated in the pathogenesis of this disease.678 Despite reports that Aβ may induce neuronal cell death through a variety of mechanisms, oxidative damage by reactive oxygen species (ROS) is thought to be one of the major causes of neuronal death induced by Aβ.9101112
NADPH oxidase (NOX), an enzyme complex composed of several subunits and a small GTPase Rac, produces superoxide radicals and mediates the intracellular oxidative stress. Subunits gp91phox and p22phox are located in the cell membrane while the other subunits, p47phox, p67phox, and p40phox are relocated to the cell membrane from the cytosol when the enzyme is activated.13 Although NOX was originally characterized as part of the host defense machinery in phagocytes, recently NOX activation has been implicated in neuronal death by oxidative stress, especially in some neurodegenerative diseases.141516 Despite many reports on NOX activation in microglia1718 or astrocytes1920 being the source of ROS involved in neuronal death induced by Aβ, the report demonstrated the activation of neuronal NOX by Aβ may also be involved.21 We investigated whether treatment with Aβ can induce activation of neuronal NOX and if this activation participates in neuronal death induced by Aβ in mixed cortical cultures.
Go to : 
MATERIALS AND METHODS
1. Mixed cortical cell cultures
Mixed cortical cell cultures, containing both neurons and astrocytes, were prepared from fetal ICR mice (Damool, Korea) at gestation days15-17 by plating dissociated cortical cells onto previously established astroglial-cell-monolayer 24-well plates (Falcon, USA).22
For astroglial beds, cortical glial cultures were prepared from postnatal ICR mice aged 1-2 days. A 1-2 day-old ICR mouse was sacrificed by cervical dislocation. The brain was excised and rinsed in cold Ca2+/Mg2+-free Hanks' balanced salt solution (HBSS), supplemented with 5 mg/mL glucose, 7 mg/mL sucrose, and 0.35 mg/mL NaHCO3. Meninges were removed under a stereoscopic microscope. The cortex was chopped into 1-2 mm3 sized pieces, put into the same solution containing 0.25% trypsin, and incubated at 37℃ for 15 min. After removal of the trypsin-containing media by a brief centrifugation for 5 min, the separated cells were moved into 1-2 mL of Eagle's minimal essential medium (MEM) containing 10% fetal bovine serum (FBS), 10% horse serum (HS), and 2 mM glutamine, and suspended vigorously 10 times up and down using a narrowed-orifice-pipet tips. Cells were plated in 24-well, multi-well plates at a density of 0.5 hemisphere/plate. The plating medium was supplemented with Eagle's MEM containing 2 mM glutamine, 10% FBS, 10% horse serum, and 10 ng/mL of epidermal growth factor in a humidified CO2 incubator (Forma, USA) containing 5% CO2 at 37℃. The culture medium was changed once a week after 14 days in vitro. Glial cultures were used for plating of the mixed cortical cell cultures between 18 and 24 days in vitro after the cell layer covers the bottom of well completely.
For mixed cortical cultures, the cells were dissected from fetal mouse brains. The fetal mice were excised by cesarean sections from a pregnant mouse on the 15-17th day of gestation under halothane anesthesia. Fetal mouse brain cortical cells were obtained using the same method described above for cortical glial cultures, and plated onto the previously established glial layer of 24-well multi-well plates at a density of 3 hemispheres/plate (approximately 2.5×105 cells per well). The plates were maintained in the same incubator. Cytosine arabinoside was added to produce a final concentration of 10 µM at 5 days in vitro, and the plates were maintained for 2 days to halt non-neuronal cell division. The culture medium was changed twice a week after 7 days in vitro. The cultures were used for the experiments at 13 or 14 days in vitro. The culture media had the same ingredients as the media, except for lower concentrations of FBS (5%) and HS (5%).
The procedures involving experimental animals complied with the regulations for the care and use of laboratory animals described by the animal ethical committee of Chonnam National University.
2. Treatment
After washing with MEM (with Earle's salts), cultures were exposed to Aβ25-35, and any drugs or siRNAs for 24 or 48 hr. Each row of the 24-well plates had 4 wells that received the same treatment. The four wells in the first row were treated with sham wash, NMDA (500 µM) was used to kill all of neurons in the second row, and the third to the sixth rows were treated with drugs or siRNAs 4 to 8 µL in each well with culture media.
3. Measurement of cell death
Cell death was morphologically assessed under a phase-contrast microscope, and a quantitative assessment of cell death was then assessed by measuring the activity of lactate dehydrogenase (LDH) in bathing media at 24 or 48 hr after treatment. The sample medium (25 µL) was taken and diluted by mixing with 125 µL of reaction buffer and mixing again with 100 µL of 0.3 mg/mL NADPH and 30 µL of 22.7 mM pyruvate solutions. Absorbance changes at 340 nm were monitored for 4 min immediately after mixing with the pyruvate solution by a microplate reader (Molecular Devices, USA). The standard enzyme was purchased from Sigma-Aldrich (USA). The data are shown as percentages (mean±SEM) of the activity values from the NMDA-treated cell group (full kill).
4. SYTOX green staining
For morphological evaluation of nuclei, SYTOX Green (Invitrogen, USA) staining was used. After fixing cells with 4% paraformaldehyde for 40 min at room temperature, the cells were permeabilized with 0.5% triton X-100 for 10 min, stained with 1 µM SYTOX Green for 15 min, and mounted with a SlowFade Antifade kit (Molecular Probes, USA). Changes in nuclear shapes and patterns were observed, and photographs were taken under a fluorescent microscope (Ex/Em=504/523 nm).
5. Measurement of intracellular ROS
Intracellular ROS induced by Aβ peptide were measured in the presence of 2,7-dichlorofluorescin diacetate (DCF-DA, Sigma-Aldrich), a fluorescent intracellular ROS indicator. Cells in the 96 well plate were incubated with 100 µL of 20 mM HEPES-buffered HBSS (pH 7.4) containing 10 µM DCF-DA and 5 mM glucose for 30 min at 37℃. Excessive dye was washed out by adding fresh culture media without phenol red, and cells were then treated with Aβ alone or in combination with apocynin, trolox, or AEBSF. After treatments, fluorescent signals for ROS generation were measured at 0.5, 1, 2, 4, 6, and 16 hr in a fluorescent multi-well plate reader (Labsystems, Finland) with excitation at 485 nm and emission at 530 nm.
6. Knockdown of gp91phox
StealthTM siRNA (Invitrogen), an siRNA set specific to gp91phox, was used. The sense and antisense strand sequences were designed as follows; sense, CCAUCCACACAAUUGCACAUCUCUU; antisense, AAGAGAUGUGCAAUUGUGUG GAUGG. For the knockdown of gp91phox expression, StealthTM siRNA of gp91phox (Invitrogen) was used. The sense and antisense sequences of gp91phox were CCAUCCACACAAUUGCACAUCUCUU and AAGAGAUGUGCAAUUGUGUGGAUGG, respectively. At 6 DIV (day in vitro), the cortical cultures were transfected with 60 pmol siRNA of gp91phox or the negative control siRNA using the GeneSilencer siRNA transfection reagent (Gene Therapy Systems, San Diego, CA).15 In brief, siRNA and GeneSilencer were each diluted to 25 µL with media and then mixed together for 10 min. Cells were fed 500 µL of fresh culture medium and overlaid with the transfection mix. After 24 hr, the cells were fed with 500 µL of culture medium. Transfected cells were used for experiments at 13-14 DIV.
7. Western blotting
To check the expression level of gp91phox, western blot analysis was performed. Cells were harvested by adding RIPA buffer (1% NP-40, 1M DTT, 100 mM PMSF, protease inhibitor cocktail), broken by an ice-cold glass homogenizer and put on ice for 30 min. After centrifugation at maximum speed for 15 min, the supernatants were assayed for protein concentration using a BCA kit. Samples containing 100 µg of protein were mixed with SDS-PAGE sample buffer, heated at 100℃ for 5 min, and loaded onto a 12% SDS-PAGE gel. After electrophoresis, the separated proteins in the gel were transferred to a nitrocellulose membrane in Towbin buffer (25 mM tris pH8.3, 192 mM glycine, 20% methanol). The membranes were then incubated with anti-gp91phox antibodies (Santa Cruz Biotechnology Inc., USA) overnight at 4℃ and developed with chemiluminescence using ECL kit (Amersham Life Science, USA). Luminescent images were analyzed with a LAS-3000 (Fujifilm, Japan).
8. Reagents
Eagle's MEM, glutamine, and HBSS were purchased from Gibco (USA), FBS and HS were purchased from Hyclone (USA). Serums were heat inactivated at 55℃ for 30 min before use. HEPES (acid), glucose, NaHCO3, NaCl, KCl, MgCl2, CaCl2, NaOH, phenol red, trypsin, cytosine arabinoside, epidermal growth factor, sucrose, Aβ25-35, ascorbate, epigallocatethin gallate (EGCG), NMDA, MK-801, and trolox were purchased from Sigma-Aldrich, Apocynin and AEBSF were purchased from Calbiochem (Germany).
9. Statistical analyses
Results are expressed as mean±SEM values, and the differences between effects were statistically evaluated using one way analysis of variance (ANOVA) followed by the Student-Newman-Keuls post hoc tests using Instat software (Graphpad Software Inc., USA). Statistical significance was accepted at p-values <0.05.
Go to :
RESULTS
Aβ25-35 is one of the β-amyloid peptide fragments known to induce neurotoxicity. Aβ25-35 did induce neuronal death in dose- and time- dependent manners (Fig. 1A). Although treatment with 10 µM of Aβ25-35 for 24 hr did not induce any significant cell death, treatment with 80 µM for 48 hr induced about 90% cell death. Chromatin condensation and nuclear fragmentation, specific features of apoptotic cells, were observed in SYTOX green-stained cells when treated with 20 µM of Aβ25-35 for 24 hr (Fig. 1B). In addition to the morphological features, treatment with anti-apoptotic drugs, such as cycloheximide (1 µg/mL, a protein synthesis inhibitor) and z-VAd-fmk (100 µM, a pan-caspase inhibitor), also significantly attenuated Aβ-induced neuronal death (Fig. 1C).
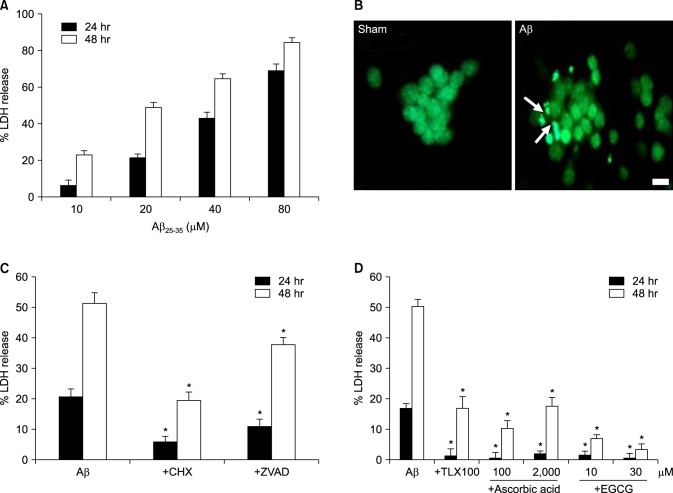 | FIG. 1β-Amyloid peptide (Aβ)25-35-induced neuronal death in mouse cortical cultures and the protective effects of antioxidants. (A) Cell death measured by assay for leaked lactate dehydrogenase (LDH) activity in the media, showing the LDH activity of the NMDA-treated cell group (% LDH release). Each column and bar are the mean±SEM from 8-20 wells. (B) Fluorescent photomicrographs from typical representative fields (200×field) of cells were taken after a 24-hour exposure to sham wash (sham) or 20 µM Aβ25-35 (Aβ). Arrows indicate fragmented and condensed chromatin stained with SYTOX green. (C) Inhibitory effect of 1 µg/mL cycloheximide (+ CHX), or 100 µM z-VAD-fmk (+ZVAD), on the 20 µM Aβ25-35-induced neuronal death after 24 and 48 hr of exposure. Each column and bar is the mean±SEM from 12-16 wells. (D) Effect of co-treatment with trolox (100 µM), ascorbic acid (100, 2000 µM) or EGCG (10, 30 µM) on the 20 µM Aβ25-35-induced neuronal death after 24 and 48 hr of exposure. Each column and bar are the mean±SEM from 8-12 wells. *Significantly different from corresponding Aβ-treated control group (p<0.05).
|
We investigated whether oxidative stress was involved in Aβ-induced neuronal death by using some well-known antioxidants. All three antioxidants used, trolox (100 µM; a vitamin E analog), ascorbic acid (100 µM, 2 mM), and EGCG (10, 30 µM) significantly inhibited neuronal death induced by the 20 µM Aβ25-35 (Fig. 1D).
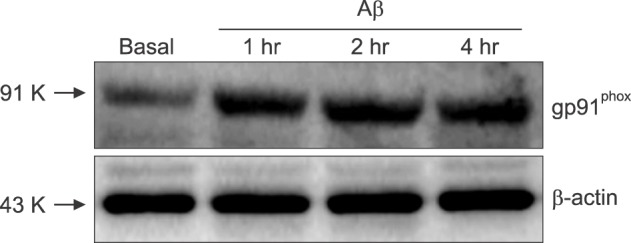
Next, we evaluated the generation of intracellular ROS in the cells treated directly with Aβ25-35, using DCF-DA, a fluorescent indicator for intracellular ROS. Fluorescent signals significantly increased in mixed cortical cultures 30 min after Aβ25-35 was added, and increased dramatically in strength thereafter to 16 hr (Fig. 2A). Furthermore, the fluorescent signals were suppressed almost completely by co-treatment with 100 µM trolox. To explore the possible role of NOX in production of intracellular ROS, we tested with AEBSF and apocynin which are known to be NOX inhibitors. The Aβ-induced fluorescent signals were suppressed almost completely by 50 µM AEBSF, similar to 100 µM trolox. Consistent effects were observed by co-treatment with 1 mM apocynin. Only a weak fluorescent signal was detected from the cells in pure astrocyte culture, and this signal was not suppressed by apocynin (Fig 2B). Furthermore, we also found that Aβ25-35 increased the expression of gp91phox, a major subunit of NOX in cells. Western blot analysis revealed that increased amounts of gp91phox were detected in the Aβ25-35-treated cells compared with the sham washed cells (Fig. 3). The elevated expression of NOX persisted for more than 4 hr after the cell was exposed to 20 µM Aβ25-35.
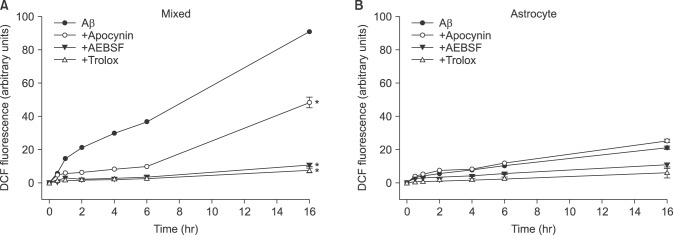 | FIG. 2Time-course of reactive oxygen species (ROS) generation by 20 µM Aβ25-35 and the effects of treatment with apocynin (1 mM), AEBSF (50 µM), and trolox (100 µM) on Aβ25-35-induced ROS generation in mouse neuron-glia mixed cortical (mixed) and astrocyte (Astrocyte) cultures. ROS generation was measured as the relative strength of fluorescence (Ex/Em=485 nm/538 nm) from the Aβ25-35-exposed cells in the presence of 2',7'-dichlorofluorescin diacetate dye. *Significantly different from negative or Aβ control groups (p<0.05).
|
As NOX inhibitors showed suppressive effects on the Aβ-induced production of intracellular ROS, we further studied the effects on Aβ-induced neuronal death. Treatment with apocynin (500 µM, 1 mM) or AEBSF (50 µM) effectively prevented cell death (Fig. 4).
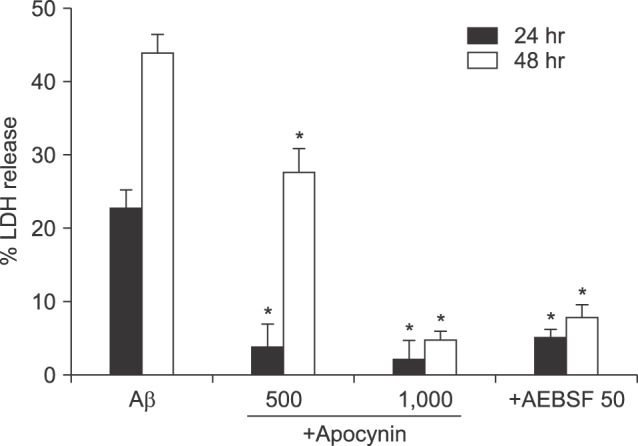 | FIG. 4Protective effects of co-treatment with apocynin (500 µM or 1 mM) or 50 µM AEBSF on 20 µM Aβ25-35-induced neuronal death after 24 and 48 hr of exposure. Cell death was measured as described in Fig. 1. Each column and bar are the mean±SEM from 8-16 wells. *Significantly different from corresponding Aβ-treated control group (p<0.05).
|
To explore more clearly the role of NOX in Aβ-induced neuronal death, gp91phox was knocked down using an siRNA-based technique. Treatment with a set of specific siRNAs successfully decreased the expression level of gp91phox (Fig. 5), and decreased the cell death induced by Aβ (Fig. 5). Comparatively, negative (mock) siRNA treatment did not show any significant effects on either the expression level of gp91phox or degree of Aβ-induced cell death.
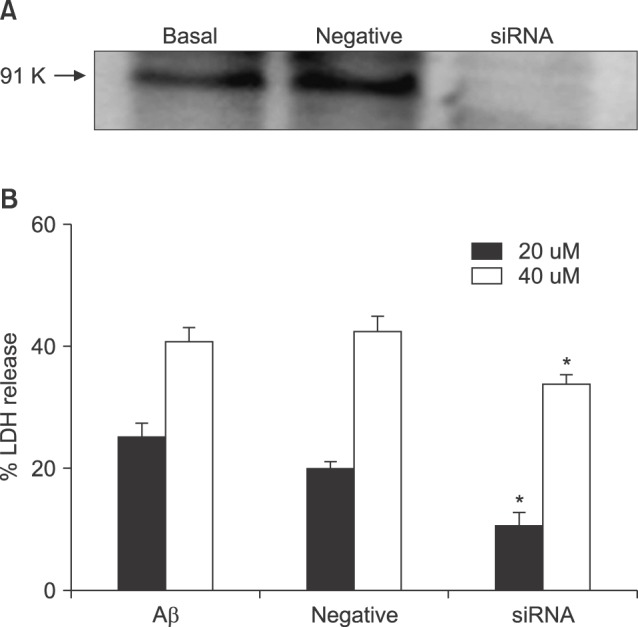 | FIG. 5Effects of knockdown of gp91phox with siRNA on 20 µM and 40 µM Aβ25-35-induced neuronal death after 24 hr of exposure. Upper representative western blots for gp91phox in sham-washed control (basal) and sister cultures 24 hr after treatment with mock siRNA (negative) and siRNA. Negative represents the scrambled mock siRNA treated group. Cell death was measured as described in FIG. 1. Each column and bar are the mean±SEM from 8-16 wells. *Significantly different from negative or Aβ control groups (p<0.05).
|
Go to :
DISCUSSION
Aggregates of Aβ peptides are the major constituent of senile plaques, the common brain lesions of Alzheimer's patients. Aβ peptides are composed of 40 (Aβ1-40) or 42 (Aβ1-42) amino acid residues.23 Aβ25-35, a β-amyloid peptide fragment, is used in many experiments as a substituent for the original Aβ peptides, because it has an equivalent neurotoxic action to the original peptides and is more easily available; however it does not exist in lesions.24
It is well established that Aβ-induced neuronal cell death is typically apoptotic.25 In the present study, we demonstrated that neuronal death is apoptotic based on morphological features, such as chromatin condensation and nuclear fragmentation, via intervention with anti-apoptotic drugs in mouse cortical cultures (Figs. 1B, C).
There are many preceding reports that oxidative stress from ROS may be involved in Aβ-induced neuronal death.910111222 The attenuation of Aβ-induced neuronal cell death by antioxidant treatment in this study indicates that oxidative stress is involved in this death process. Furthermore, we demonstrated that a lot of ROS were produced in the Aβ25-35-treated cells, persisting for more than 4 hr, using a fluorescent indicator for intracellular ROS, DCF-DA (Fig. 2A). Increased ROS production by Aβ25-35 occurred only in the mixed cultures with few signals from the pure astrocyte cultures (Fig. 2B). These results suggest that Aβ25-35 mainly produces ROS from neurons in mixed cortical cultures. NOX is distributed in a wide range of central nervous systems (CNS),2627 and is found in most CNS cells, such as astrocytes, microglia, and neurons.1718192021 In contrast to the present study, in a study of rat hippocampal neuron-astrocyte co-culture system, Abramov et al.19 reported that NOX was involved in Aβ-induced ROS generation and neuronal death, and that ROS generation occurred in astrocytes, but not in neurons. We cannot conclude whether this difference came from the difference in species (mouse vs rat) or tissues (cortex vs hippocampus).
Next, we tried to delineate the mechanism of ROS generation inside cells treated with Aβ25-35. We found that the expression level of gp91phox was increased following Aβ25-35 treatment by western blot analyses (Fig. 3). Treatment with known NOX inhibitors such as apocynin28 and AEBSF,29 not only reduced intracellular ROS production, but also attenuated neuronal death induced by Aβ25-35, (Fig. 4). In this study, AEBSF completely blocked ROS production in both mixed cultures or pure astrocyte cultures, but apocynin only partially reduced the ROS in co-cultured cells, with no effect on pure astrocytes. These data suggest the possible involvement of certain serine proteases in Aβ-induced ROS generation and cell death, considering that AEBSF is a known inhibitor of serine proteases.30
To confirm the roles of NOX in neuronal death induced by Aβ25-35, we introduced a set of gp91phox-specific siRNA into the cells; siRNAs effectively reduced both the expression of gp91phox and Aβ25-35-induced cell death (Fig. 5).
Taken together, the above results suggest that NOX is involved in Aβ-induced ROS generation and neuronal death in mixed cortical cultures.
Go to :
 XML Download
XML Download