

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hypertension accelerates atherosclerosis and can induce vascular inflammation mediated by various mediators. 12 Plaque progression is associated with the action of angiotensin II and inflammatory and vascular smooth muscle cells, and oxidative stress and endothelial dysfunction, in patients with hypertension.34
Intravascular imaging systems, especially intravascular ultrasound (IVUS), have been used to evaluate the mechanisms of plaque progression or regression,56 and several studies have shown the effects of statins on the course of coronary atherosclerosis and the relationship between concentrations of low-density lipoprotein cholesterol (LDL-C) with statin treatment and plaque progression or regression.78
A previous study demonstrated that an increase in systolic blood pressure at follow-up was independently associated with plaque progression in patients with very low achieved LDL-C levels.9 However, the factors related to plaque progression are not well understood in statin-treated patients with angina pectoris and hypertension. Therefore, the aim of this study was to evaluate the predictors of plaque progression by use of virtual histology (VH)-IVUS in statin-treated hypertensive angina patients whose achieved LDL-C level was <70 mg/dL at follow-up.
MATERIALS AND METHODS
1. Patient population
This study was a retrospective, single-center study. A total of 78 statin-treated hypertensive angina patients who achieved an LDL-C level of <70 mg/dL with statin treatment and who underwent gray-scale and VH-IVUS in non-intervened coronary segments were identified from the Chonnam National University Hospital VH-IVUS registry database. Follow-up angiogram and IVUS examinations were performed in patients with chest pain or a positive finding on a stress test as well as in asymptomatic patients during routine follow-up. All patients received 10 mg/day of rosuvastatin from baseline to follow-up.
The effects of 9 months of rosuvastatin therapy on coronary plaque progression were evaluated by using VHIVUS. The included patients were divided into plaque progressors (n=30) and plaque regressors (n=40) according to the baseline minimum lumen area (MLA) site at the 9-month follow-up.
Chronic kidney disease (CKD) was defined as estimated creatinine clearance (CrCl) <60 mL/min by use of the Cockcroft-Gault formula10: CrCl = [(140-age)×mass (in kg) {×0.85 if female}/72×serum creatinine]. Patients on dialysis were not included in the present study.
The presence of stable angina was determined according to the Canadian Cardiovascular Society classification. The presence of unstable angina was determined by chest pain within the preceding 72 h with or without ST-T-wave changes or positive cardiac biochemical markers (creatine kinase-myocardial band or cardiac-specific troponin-I).
The study protocol was reviewed and approved by the institutional review board of Chonnam National University Hospital, Gwangju, Korea (CNUH-2013-054). Hospital records of all patients were reviewed to obtain clinical demographics and medical history. Written informed consent was obtained from all patients before cardiac catheterization.
2. Laboratory analysis
Peripheral blood samples were centrifuged, and serum was removed and stored at -70℃ until the assay could be performed. Apolipoprotein (apo) B and apoA1 were analyzed immunoturbidometrically (Orion Diagnostica, Espoo, Finland). The details of other lipid determination methods have been previously published.11
3. Quantitative coronary angiography
The target lesion was an intermediate lesion with diameter stenosis between 30% and 60% that was not treated by percutaneous coronary intervention. The coronary angiogram was analyzed with the validated quantitative coronary angiography system (Allura DCI program, Philips Medical Systems, the Netherlands).12 With the outer diameter of the contrast-filled catheter as the calibration standard, the minimal lumen diameter and reference diameter were measured in diastolic frames from orthogonal projections.
4. Gray-scale and VH-IVUS analysis
All pre-intervention IVUS examinations were performed after intracoronary administration of 300 µg of nitroglycerin. A 20-MHz, 2.9-Fr IVUS imaging catheter (Eagle Eye, Volcano Corp, Rancho Cordova, CA, USA) was advanced >10 mm beyond the lesion, and automated pullback was performed to a point >10 mm proximal to the lesion at a speed of 0.5 mm/s.
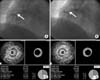
A representative case of plaque progression is shown in Fig. 1. Quantitative volumetric IVUS analyses were performed across the entire lesion segment, and cross-sectional analysis was performed at the MLA site. Quantitative volumetric gray-scale IVUS analysis was performed according to the American College of Cardiology Clinical Expert Consensus Document on Standards for Acquisition, Measurement and Reporting of Intravascular Ultrasound Studies.13 Measurements were made at every 1-mm interval for the region of interest, which was defined as the segment between distal and proximal reference sites. Volumetric data were generated by the software by using Simpson's method. The external elastic membrane (EEM) and lumen cross-sectional areas (CSAs) were measured. Plaque plus media (P&M) CSA was calculated as EEM minus lumen CSA, and plaque burden was calculated as P&M divided by EEM CSA. Total atheroma volume (TAV) was determined by summation of the plaque area, which was defined as the difference between EEM and lumen area, for all evaluable images as TAV=Σ (EEM area - lumen area). The percent atheroma volume (PAV) was calculated as the percentage of the sum of EEM areas occupied by TAV: PAV=100 × [Σ (EEM area - lumen area)/Σ (EEM area)]. Soft plaque was less bright compared with the reference adventitia. Fibrotic plaque was as bright as or brighter than the reference adventitia without acoustic shadowing. Atheromas have been classified in four categories by gray-scale IVUS: (1) soft plaque (lesion echogenicity less than the surrounding adventitia), (2) fibrous plaque [intermediate echogenicity between soft (echolucent) atheromas and highly echogenic calcified plaques], (3) calcified plaque (echogenicity higher than the adventitia with acoustic shadowing), and (4) mixed plaques (no single acoustical subtype represents >80% of the plaques). VH-IVUS analysis classified the color-coded tissue into four major components: (1) dark green (FT), (2) light green (FF), (3) red (NC), and (4) white (DC).14 Two experienced, independent IVUS analysts defined thin-cap fibroatheroma as a lesion fulfilling the following criteria in at least three consecutive frames: NC≥10% without evident overlying fibrous tissue and PAV≥40%. VH-IVUS analysis was reported in absolute amounts and as a percentage of plaque area or volume.
5. Statistical analysis
The Statistical Package for the Social Sciences (SPSS) for Windows, version 15.0 (Chicago, IL, USA), was used for all analyses. Continuous variables were presented as the mean value±1 SD; comparisons were conducted by Student's t-test or nonparametric Wilcoxon test if the normality assumption was violated. Discrete variables were presented as percentages and relative frequencies; comparisons were conducted by chi-square statistics or Fisher's exact test as appropriate. Multivariate analysis was used to identify the independent predictors of plaque progression at the baseline MLA site from baseline to follow-up. All variables with a p value<0.10 in the univariate analysis were tested. A p value<0.05 was considered statistically significant.
RESULTS
The baseline characteristics of the patients and their coronary angiographic findings are summarized in Table 1. The prevalence of unstable angina (vs. stable angina), current smoking, and CKD was higher in progressors than in regressors. Prior percutaneous coronary intervention had been performed more frequently in progressors than in regressors. Baseline creatinine was significantly higher and CrCl was significantly lower in progressors than in regressors. Baseline apoB and apoB/A1 were significantly higher in progressors than in regressors. No significant difference was observed in the use of RAS blocker between the groups. The target lesion was located in the right coronary artery more frequently in progressors than in regressors.
Baseline gray-scale and VH-IVUS findings are summarized in Table 2. There were no significant differences in baseline gray-scale or VH-IVUS findings between progressors and regressors.
Follow-up clinical, laboratory, gray-scale, and VH-IVUS data are summarized in Table 3. The mean follow-up duration from baseline to follow-up IVUS examinations was 9.2±2.5 months in progressors and 9.0±1.8 months in regressors. No significant difference was observed in the use of RAS blocker between the groups. Follow-up plaque burden at the MLA site was significantly greater in progressors than in regressors. Absolute and relative FT areas at the MLA site were significantly greater in progressors than in regressors.
Data for changes in laboratory, gray-scale, and VH-IVUS findings from baseline to follow-up are summarized in Table 4. EEM CSA, P&M CSA, and plaque burden at the MLA site increased in progressors but decreased in regressors from baseline to follow-up. Absolute and relative FT areas at the MLA site increased in progressors but decreased in regressors from baseline to follow-up. EEM volume and TAV increased in progressors; by contrast, EEM volume, TAV, and PAV decreased in regressors from baseline to follow-up. Absolute FT volume increased in progressors but decreased in regressors from baseline to follow-up.
Independent predictors of plaque progression at the MLA site are shown in Table 5. The following variables were tested to determine the independent predictors of plaque progression at the baseline MLA site (variables with p<0.10 in the univariable analysis: CKD, baseline apoB/A1, smoking, right coronary artery lesion, systolic blood pressure, ΔEEM CSA at MLA site, and Δrelative FT area at MLA site). CKD [odds ratio (OR): 2.13, 95% confidence interval (CI): 1.77-2.53, p=0.013], apoB/A1 (OR: 1.25, 95% CI: 1.12-1.40, p=0.023), and smoking (OR: 1.76, 95% CI: 1.23-2.22, p=0.038) were independent predictors of plaque progression at follow-up.
DISCUSSION
The present study demonstrated that 1) there was a higher prevalence of current smoking and CKD and a higher baseline level of apoB/A1 in progressors than in regressors; 2) EEM CSA, P&M CSA, and plaque burden at the MLA site increased in progressors and by contrast decreased in regressors from baseline to follow-up; 3) absolute and relative FT areas at the MLA site increased in progressors but decreased in regressors from baseline to followup; and 4) CKD, smoking, and baseline apoB/A1 were associated with plaque progression in patients with angina pectoris and hypertension after moderate-intensity treatment with rosuvastatin.
Hypertension is associated with progression of atherosclerosis by the action of angiotensin II, vascular smooth muscle cells, and oxidative stress and endothelial dysfunction. 12 Reductions in LDL-C levels produced by statins are associated with coronary plaque regression.78 Lee et al.15 reported that statin therapy significantly reduces the risk of the composite of 1-year major adverse cardiac events including death, recurrent myocardial infarction, target vessel revascularization, and coronary artery bypass grafting in patients with acute myocardial infarction who have baseline LDL-C levels <70 mg/dL. Bayturan et al.9 reported that an increase in systolic blood pressure at follow-up is one of the risk factors for plaque progression despite the achievement of very low levels of LDL-C. However, the predictors of plaque progression in statin-treated hypertensive angina patients who achieve LDL-C levels <70 mg/dL are not well known.
In the present study, CKD was shown to be an independent predictor of plaque progression in patients with angina pectoris and hypertension who achieved very low LDL-C levels after moderate-intensity rosuvastatin treatment. CKD is associated with poor clinical outcomes,16 increased prevalence of coronary artery disease,17 and accelerated atherosclerosis.18 Renal dysfunction is associated with atheroma progression and an unstable plaque phenotype, which provides novel insight into the mechanisms of poor clinical outcome in CKD patients. The exact mechanisms of plaque progression and formation of vulnerable plaque in CKD patients are not well known, but several mechanisms have been postulated. CKD patients have elevated serum matrix metalloproteinases that are associated with plaque growth and fibrous cap disruption.19 Matrix metal-loproteinases are related to endothelial dysfunction, vascular remodeling, and arterial stiffening in CKD patients.20 CKD potentiates foam cell formation, which enhances macrophage infiltration into the vascular wall and inhibits cholesterol efflux.21 Therefore, aggressive statin therapy is needed to regress and stabilize coronary plaque in CKD patients.
In the present study, smoking was also shown to be independently associated with plaque progression in patients with angina pectoris and hypertension who achieved very low LDL-C levels after statin treatment. Lehman et al.22 reported that smoking was associated with plaque progression in patients who presented to the emergency department with acute chest pain but who initially showed no evidence of acute coronary syndromes by contrast-enhanced 64-slice computed tomography. Epidemiological studies of diverse populations have shown that smoking is a risk factor for the development of coronary and peripheral vascular disease.23 Therefore, smoking cessation is needed to improve the lipid profile and reverse the atherogenic effects of smoking.
Last, the baseline apoB/A1 concentration was an independent predictor of plaque progression at follow-up in statin-treated patients with angina pectoris and hypertension in the present study. Juonala et al.24 reported that apoB and apoB/A1 are directly related to adulthood carotid artery intima-media thickness and that apoB/A1 is superior to the ratio of LDL-C to high-density lipoprotein-cholesterol (HDL-C) in predicting increased carotid artery intima-media thickness in adulthood. ApoB-containing lipoproteins carry lipids from the liver and intestine to the sites of use, whereas apoA1-containing particles mediate the reverse cholesterol transport and bring excess cholesterol from peripheral tissues to the liver.25 Concentrations of apoB and apoA1 reflect the particle numbers of their respective lipoprotein classes and thus the opposite aspects of the risk. A high apoB/A1 indicates the number of atherogenic lipoprotein particles that are likely to be deposited in the arterial wall.26 ApoB/A1 is associated with plaque progression even if very low levels of LDL-C are achieved after statin treatment.
There are several limitations to mention. First, the analyses were retrospective and are subject to the limitations inherent in this type of clinical investigation. Second, the results of this study are only applicable to selected hypertensive angina patients. So, the results in the present study might not be generalizable in all patients with coronary artery disease such as acute myocardial infarction. Third, the prevalence of unstable angina (vs. stable angina) was significantly higher in progressors than in regressors. However, the plaque characteristics were not significantly different between progressors and regressors and unstable angina itself was not an independent predictor of plaque progression at follow-up. Fourth, the prevalence of CKD was significantly higher in progressors than in regressors. Because it is possible for CKD patients to have a large amount of calcium compared with non-CKD patients, they might be resistant to regression by statin therapy. Fifth, we defined decreased renal function on the basis of estimated creatinine clearance rather than direct measurements of the glomerular filtration rate. Sixth, we did not have information about the duration of exposure and the amount of smoking. Seventh, there is a possibility of over-estimation of the NC component in lesions with heavily calcified plaques by VH-IVUS.
Clinical factors including CKD, smoking, and apoB/A1 rather than plaque components detected by VH-IVUS are associated with plaque progression in hypertensive angina patients who achieve very low LDL-C after statin treatment.





 XML Download
XML Download