

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Aging is a cumulative effect of various deterioration processes, such as oxidation, telomere curtailment, fraying and tearing, and disease-specific processes such as accumulation of bone cell oxidation (osteoporosis) or accumulation of blood vessel deposits (atherosclerosis). However, the exact molecular mechanism behind age-related diseases has yet to be established.
Osteoporosis and cardiovascular disease (CVD) are two common conditions in postmenopausal women. Both diseases are becoming progressively widespread as the population ages. Thus, aging and osteoporosis are intimately linked.1 Similarly, the incidence of CVD is low in premenopausal women but increases steadily in postmenopausal women. Osteoporosis is commonly referred to as the "silent thief," because there are no noticeable symptoms associated with its progression until it manifests in the form of a fragility fracture. A similar analogy can be made for CVD, which also acts like a "silent thief." According to Kang2, CVD and osteoporosis undoubtedly have a similar basis of oxidative stress as for many other diseases. In fact, the etiology of a great many maladies has been found to be associated with inflammation and immune dysregulation related with natural aging.3 With the widespread incidence of postmenopausal osteoporosis (PMOP) in the aging population, the impact of this condition on public health is becoming increasingly critical.
Primary osteoporosis is classified into two subtypes: (a) type I osteoporosis, a general bone disease in postmenopausal women resulting from estrogen deficiency after menopause, and (b) type II osteoporosis (also known as senile osteoporosis or age-related osteoporosis), which is related to aging in men and women.4 The estrogen deficiency is primarily caused by enhanced osteoclastic and depressed osteoblastic activity; especially, early postmenopausal women experience an accelerated bone loss, with an average bone mineral loss of 0.5-2% per year, which then gradually slows down.2
Irrespective of gender, estrogen is important in bone homeostasis and skeletal growth. As a consequence of declining estrogen, women after menopause undergo rapid bone damage.5 It has been suggested that estrogens have vasoprotective effects and reduce cardiac events; hence, estrogens are used as a medication for the management of postmenopausal bone damage and cardiac events. Owing to its ability to regulate the immune system and oxidative stress, estrogen has been found to regulate body homeostasis; such regulation is also done by estrogen's direct effects on target cells.3,4,5 Although the specific molecular etiology has yet to be shown, vascular calcification is a result of a dysregulated process and has been shown to recapitulate processes similar to bone formation.
In this review, we attempted to explore the similarities in the mechanisms proposed for PMOP and CVD with a gleaning of the clinical information that supports the interplay between these situations.
BONE REMODELING AND PATHOPHYSIOLOGY OF OSTEOPOROSIS
The mineralized skeleton is formed of a protein matrix mostly composed of collagen type I fibers oriented in linear tufts. After achieving peak bone size and mineral density through bone modeling in early adulthood, human bones undergo a renovation event known as remodeling. During bone remodeling, osteoclasts remove damaged or old bones, which are consequently replaced with new bone generated by osteoblasts, and this entire physiological event takes place within a period of approximately 6 months.3 Without any change (loss or gain) of bone mass, the primary function of bone remodeling is to repair bone. Theoretically, each year through bone remodeling, about 25% of trabecular bone and 3% of cortical bone is changed. There is a homeostasis in terms of bone remodeling between osteoblastic bone production and osteoclastic bone destruction.6
OSTEOCLASTS AND THE RECEPTOR ACTIVATOR OF THE NF-κB/ RANK LIGAND/ OSTEOPROTEGERIN SYSTEM
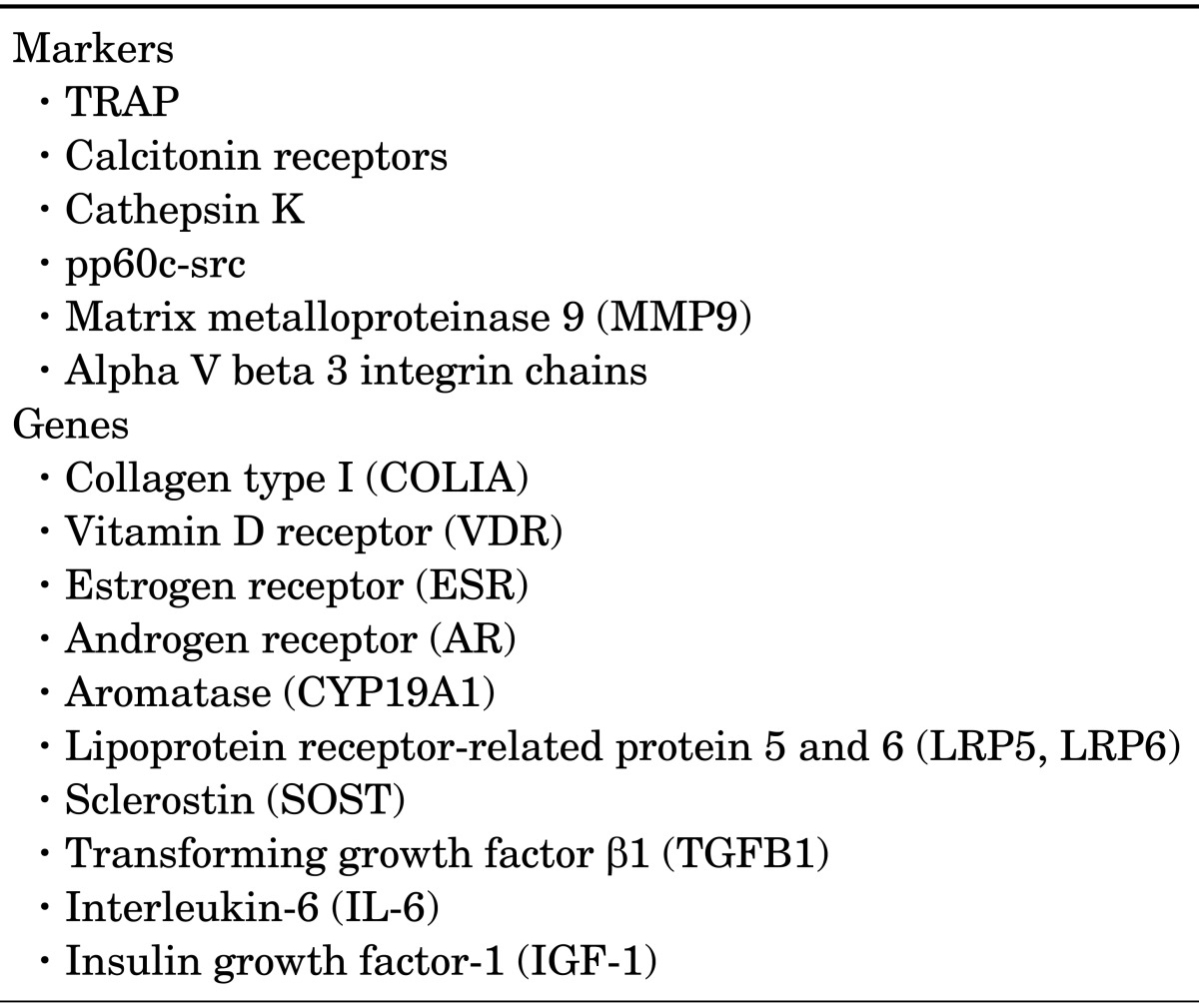
Osteoclasts are the unmatched cells of the body, employed in resorbing bone (Table 1). These giant multinucleated cells create a sealing zone by binding to bone surfaces and develop a ruffled-membrane border at the bone resorption site for facilitating bone removal by increasing the cell surface area for secretion. Formation of hydrochloric acid at the ruffled-membrane border reduces bone minerals. The exposed collagen matrix is damaged by acid-resistant endosomal and lysosomal enzymes. Finally, bone mineral and cleaved collagens are released into the general circulation through extracellular circulation, thus completing the process of bone resorption.3,7
The osteoclast precursors, circulating in the macrophagemonocyte cells, first transform to pre-osteoclasts, which finally integrate together to shape huge mature osteoclasts.3 Osteoclast differentiation factor (ODF) and osteoprotegerin (OPG) are similar in structure and function and both promote osteoclast formation. The osteoclast formation occurs through the dispensation of the monocytic survival factor macrophage colony stimulating factor (M-CSF).8
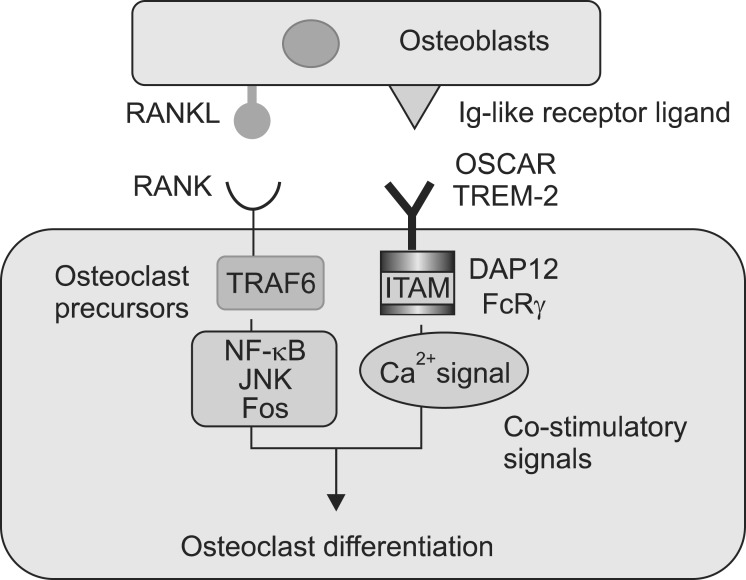
Osteoclast proliferation is mediated by the receptor activator of NF-κB (RANK). RANK is an osteoclast receptor, combined with the receptor activator of NF-κB ligand (RANKL) and an osteoblast cytokine, along with many other proteins. Currently, in bone science, RANKL is used to define the osteoclastogenic cytokine, whereas OPG and RANK are used to define its inhibitor and the receptor for RANKL, respectively. It is now clear that RANKL is the key final effector osteoclastogenic cytokine and initiates osteoclast differentiation in the presence of M-CSF (Fig. 1).8 OPG inhibits the association of RANKL with the RANK receptor, and thus acts as a trap receptor; it moderates osteoclastogenesis and bone resorption. In addition, OPG captures RANKL before it gets linked to RANK, thus suppressing bone resorption.9
Inflammatory processes have been found to be responsible for making the human body prone to osteoclastic bone loss. This further indicates the involvement of inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), interleukin (IL)-1, and IL-6 in osteoclast formation.10 IL-1 and TNF-α have long been suspected for their role in osteoclast formation, especially in PMOP. It is also now known that cytokines such as NF-κB, IL-1, and M-CSF (which have long been implicated in osteoclastic destruction) act by increasing RANKL formation, thus promoting their susceptibility to prevailing RANKL concentrations.11
The US Food and Drug Administration has approved the drug denosumab directed against RANKL for fracture prevention and amelioration for managing postmenopausalrelated osteoporosis and other forms. In addition to calcineurin- NFATc1, calcium signaling actuates the CaMKCREB protein pathway, indicating its involvement in osteoclastogenesis.3
Our current understanding of the pathophysiology of vascular and cardiac valve calcification and osteoporosis indicates that a common pathogenetic mechanism is shared by both processes. It has been found that both the bone and the cardiovascular mechanism involve common factors such as proteins (cathepsins, bone morphogenetic proteins, matrix Gla protein, receptor activator of RANKL, and osteoprotegerin), phosphate, parathyroid hormone (PTH), oxidized lipids (ox-lipids), and vitamins K and D. The correlation between osteoporosis and vascular/valvular calcification and the improved risk of cardiovascular disorders in patients with osteoporosis have also been confirmed by various clinical studies.8,9,10,11,12
AGE AND BONE REMODELING
In the aging skeleton, the amount of defective osteoblasts depends on multiple factors: (i) a reduction in the amount of mesenchymal stem cells, (ii) imperfect differentiation/ proliferation of progenitor cells or detour of these progenitors on the way to the adipocyte origin, and (iii) increased apoptosis. On the other hand, the amount of osteoclasts in cancellous bone gradually diminishes with age along with an accompanying reduction in RANKL levels in the bone marrow plasma. Enhancement in osteocyte apoptosis with age has also been found to be associated with decreased levels of sclerostin protein in bone and RANKL.13
With the diminished level of estrogen at menopause, the resorption cavities increase with increases in bone resorption. Insufficiency in bone production leads to imperfect refilling of resorption cavities with new bone causing a persistent decrease of bone mass. The reactive oxygen species (ROS) created in the reaction of tartrate-resistant acid phosphatase (TRAP) (found on the surface of osteoclasts) with hydrogen peroxide degrades collagen and other proteins. In addition, vitamin D also has a direct nongenomic impact on the formation of ROS. Owing to the antioxidant property of estrogen, it inhibits the generation of such reactive species. While PTH and vitamin D stimulate bone resorption by stimulating ROS production in osteoclasts, calcitonin has inhibitory effects on bone resorption by inhibiting ROS production.4,11
OXIDATIVE STRESS AND BONE REMODELING
The biochemical link between bone metabolism and oxidative stress has been established by a number of studies. Oxidative stress is described as an imbalance between antioxidants and prooxidants in favor of the former, potentially leading to injury. With low antioxidant levels, the markers of oxidative damage increase.14,15,16 By promoting antioxidant defense, the cells, under normal physiological circumstances, can quench free radical attacks or oxidative stress. The human body is equipped with a number of endogenous antioxidant protection mechanisms, including the endogenous antioxidant enzymes glutathione peroxidase (GPx), catalase (CAT), superoxide dismutase (SOD), and metal chelating proteins. In addition, the body also possesses an alternative mechanism in the form of repair antioxidants, such as lipase DNA repair enzymes, transferase, and protease, which take care of oxidative damage when endogenous antioxidants (e.g., uric acid) or antioxidants derived from the diet fail to prevent such damage. Despite all the preventive mechanisms, when antioxidants fail to control the oxidative damage, diseases such as CVD and osteoporosis develop.17,18
ROS have been found to contribute to the aging process as well as to pathological conditions such as PMOP and atherosclerosis. It has already been confirmed that oxidative stress, improved by ROS, increases the rate of bone loss; hence, oxidative stress is a risk factor for osteoporosis.17
ROS, generated in oxidative processes, are at tolerably low levels in cells. However, the intracellular sites, including nuclear membranes, endoplasmic reticulum, and mitochondria are the major sources for the formation of ROS by way of electron transport chains. Mitochondrial metabolism and ROS formation are also responsible for the bone osteoclastic activity and the bone mass loss induced by estrogen deficiency in PMOP.14
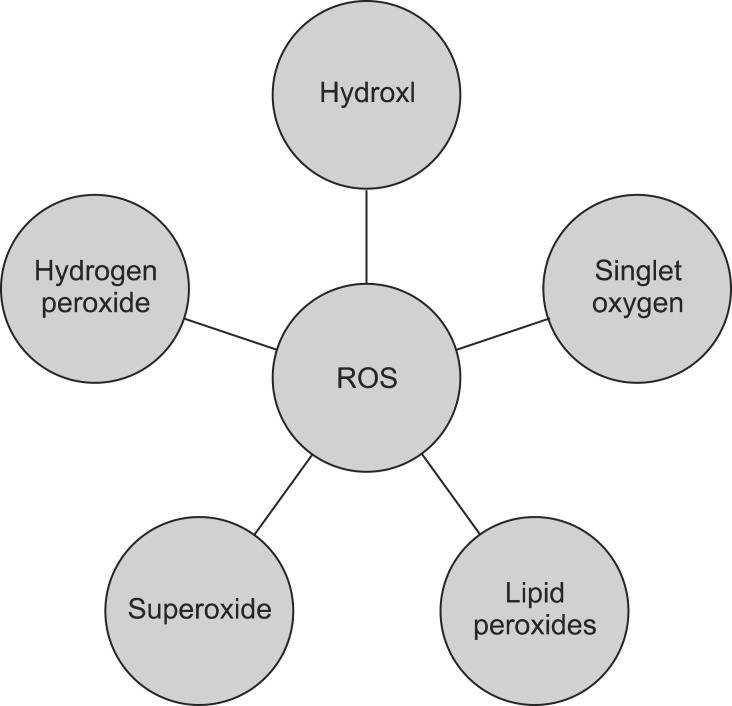
ROS have extremely reactive chemical interactions, and when they exist in excess, they repress the protective systems, resulting in cell detriment and lipid peroxidation. Singlet oxygen, hydroxyl radicals, hydrogen peroxide, superoxide radicals, and lipid peroxides are oxygen-containing molecules and free radicals. With age, ROS production increases, causing several chronic diseases including osteoporosis (Fig. 2).15,16,17,18,19
The role of free radicals in bone metabolism differs between pathological and normal physiological situations. In physiological situations, the formation of ROS by osteoclasts helps in bone remodeling by increasing the demolition of calcified tissue.20 ROS not only modulate osteoclastic transformation of bone progenitors, they also modulate osteoblastic transformation. In contrast, oxidative stress can obstruct bone cell transformation of the progenitor osteoblast cell line and of a marrow stromal cell line that undergoes osteoblastic transformation.20 Hydrogen peroxide modulates intracellular calcium (Ca2+) metabolism in osteoblasts by improving Ca2+ liberation from the intracellular Ca2+ depots.21 In osteoblasts, hydrogen peroxide also reduces cell growth, alkaline phosphatase (ALP) enzyme activity, mineralization, calcification, and gene expression of osteogenic markers such as bone sialoprotein, ALP, and runt-related transcription factor 2.22
AGING AND FUNCTION OF OXIDIZED LIPIDS AND LIPOPROTEINS IN PMOP
Both human and animal studies show that aging and the related increase in oxidative stress drive bone loss.1 Despite significant advances in medicine, osteoporosis and CVD together are still responsible for most of the morbidity and mortality in elderly persons.23
Monocyte chemotactic factors (M-CF) are strong stimulants of osteoclastic transformation and oxidized lipids induce the endothelial expression of M-CF. Thus, oxidized lipids would be anticipated to improve bone resorption by restoration and transformation of osteoclast pioneer cells. Osteoporosis is partially a result of reduced bone production by osteoblasts with aging.23
Bone and vascular tissue are known to share several features at both the molecular and the cellular level. Bone and marrow include counterpart cells in the artery wall, which are endothelial cells, progenitor osteoblast cells, and monocyte- derived osteoclasts. Both atherosclerotic arteries and bone include common molecules such as matrix Gla protein, osteopontin, collagen I, bone morphogenetic protein, osteocalcin, osteonectin, matrix vesicles, and nitric oxide.24
Lipoproteins and lipids accumulate in bone and undergo oxidation, probably affecting the cellular molecules of osteoblastic cells in bone; they obstruct appropriate boneproducing activity, similar to atherosclerosis.24 This can be attributed to the fact that bone includes an important amount of blood vessels, with cellular components of bone placed in proximity to the intertwined vascular beds. The gathering of lipids in the tissues of osteoporotic bone has been shown.20,21,22,23,24 In addition, lipids have also been shown to gather in osteon of mice and near bone vessels in osteoporosis. Given that the immature osteoblasts are placed immediately contiguous to the subendothelial region of bone vessels, lipid accumulating in the subendothelial region would be anticipated to obstruct transformation of the bone-producing cells.23,24,25
Highly reactive ROS molecules can assault almost all cell molecules. Lipid peroxidation may have an important role in bone cell and tissue metabolism. The obstruction of osteoblastic transformation has been observed on treatment with oxidized lipids.24,25 Studies have observed that administration of antioxidants dissolves osteocyte and osteoblast apoptosis in the bone of ovariectomized or aged mice.23,24,25,26 This suggests that oxidative stress might decrease the lifespan of osteoblasts and osteocytes.
PARAOXONASE (PON): A WAY FROM THE HDL TO THE BONE
There are common risk factors for developing conditions like CVD and POMP, and researchers have already established an association between CVD and PMOP. Serum PON1 has been found to be directly involved in the pathogenesis of atherosclerosis. "Healthy HDL" is armed with active PON1 enzyme, for which the main function is to suppress the formation of oxidized lipids and lipoproteins. On the other hand, "dysfunctional HDL" reduces PON1 activity, which potentially results in increasing the formation of oxidized end products, which in turn, activate lectin-like oxidized LDL receptor-1 (LOX-1). LOX-1, an oxidized LDL receptor expressed in vascular endothelium and a multiligand receptor, has been implicated in endothelial dysfunction and atherosclerosis.27 Therefore, this suggests that oxidized lipids play a role in vascular calcification and osteoporosis. PON1 shows peroxidase-like activity and reduces lipid hydroperoxides to hydroxides; both hydroperoxides and hydroxides are ROS produced under oxidative stress.1,27 Due to the presence of PON1, HDL shows antioxidant properties and reduces the accumulation of lipid peroxidation products. These lipid peroxides are unstable and are produced from polyunsaturated fatty acids; the dissociation of lipid peroxides yields a complex series of compounds. The mechanism of cellular damage in humans involves lipid peroxidation and is used as an indicator of oxidative damage in cells and tissues.28,29 There is also evidence showing the role of lipid oxidation in developing osteoporosis.30 Oxidation of lipids is linked to atherosclerosis, and epidemiologic evidence shows the association between atherosclerosis and osteoporosis.23,24,25,26,27
In 1953, a protein was identified as an organophosphatehydrolyzing enzyme in mammalian tissue; thus, PON1 in serum was discovered.29 PON1 hydrolyzes the toxic metabolite of parathion, paraoxon, and also shows arylesterase activity.28 The preferred natural substrate of paraoxonase has yet to be shown. Enzymatic characterization of PONs shows lactones to have substrate specificity. The naturally occurring homocysteine-thiolactone may be the endogenous substrate for PON.31
PON has three members, which are located on chromosome 7: PON1, PON2, and PON3. PON1 and PON3 are detectable in serum as bound to high-density lipoprotein (HDL) and are expressed in the liver. PON2 is not detectable in serum, but is found in various tissues, including the kidney, testis, liver, and brain.28,29 The amino acid similarity of PON1, 2, and 3 is about 65%. Because of high similarity, these enzymes may possess similar biochemical function.27,28,29,30,31 All three members of the PON family have been shown to prevent cell-mediated oxidative transformation of LDL.28 The increased production of mitochondrial ROS can be induced, for example, by oxidized LDL.24 In vitro research has shown that lipid oxidation products are found to improve osteoblastic transformation of vascular cells in vitro, but they obstruct such transformation of bone cells.23,24,25,26,27 Moreover, oxidation products of LDL enhance the osteoporotic decrease in bone by inducing progenitor marrow stromal cells to undergo adipogenic rather than osteogenic transformation.26,27,28,29,30 Previously, we showed that a PMOP study group had decreased total antioxidant status, whereas the increased total peroxidase was different from that in the non-osteoporosis group.1 This indicates that lipid peroxidation might have a significant role in the pathogenesis of osteoporosis and CVD.31 In order to inhibit lipid peroxidation, antioxidant therapy would be effective, and PON1 being an antioxidant agent, pharmaceutical agents that increases PON1 might be a therapeutic option in PMOP.1,31,32
About 200-odd one-nucleotide polymorphisms (ONPs) in PON1 at diverse regions of the gene were identified during the cloning of the gene in 1993.28 The reduction of oxidized LDL-catalyzed PON1 might affect bone remodeling, and the possible correlations of these ONPs to bone mineral density have also been described. In postmenopausal women, the SNPs of PON1 and PON2 have been found to be related with bone mineral density for the femoral neck or lumbar spine, and the 584GG, 172TT, and 959CC genotypes represent risk factors for decreased bone mass. However, the molecular mechanism underlying the association of ONPs of PON with bone mineral density has yet to be clarified.33
Apart from stimulating apoptosis of osteoblastic cells, oxidized LDL also inhibits bone morphogenetic protein-2-induced osteoblast transformation trough oxidative stressindependent mechanisms. PON1 can prevent these effects of oxidative stress on bone formation. The PON1 gene has been implicated in reducing bone mineral density in bone fragility cases. Moreover, oxidized LDL also inhibits osteoblastic differentiation in bone and bone osteoblasts.30,31,32,33
Oxidized lipids are suspected to have adverse impacts on bone by targeting osteoclastic cells (increased osteoclastic activity),34 whereas PON1 preserves LDL and HDL from oxidation triggered by either a free radical procreator or copper ion; reduced sera PON1 activity has been observed in patients with osteoporosis in comparison to healthy persons.35 However, the mechanism of this reduction in sera PON1 enzyme activity in patients with osteoporosis remains unclear. This reduction could be associated with increased lipid peroxidation, because of inhibition of PON1 activity by oxidized lipids.31 Protection against oxidized LDL can be achieved through PON1 inactivation, and PON1 inactivation occurs through the reaction between the free sulfhydryl group of PON1 and particular oxidized lipids in oxidized LDL. Studies have shown that oxidized lipids induce the expression of cytokines.27 Interleukins are known to down-regulate serum PON1 activity. Direct reactions with the cells through receptor-mediated responses or via the formation of other inflammatory agents such as cytokines might cause the observed impacts of the oxidized lipids on bone cells.33,34,35,36 Therefore, a reduction in PON1 in serum of patients with osteoporosis could be associated with the increase of those cytokines.36 Owing to estrogen deficiency in the postmenopausal period, a higher prevalence of increased serum IL-17A levels has been observed. Such enhancement in IL-17A might induce chronic inflammatory events, causing CVD and osteoporosis. In addition, as part of the inflammatory response, PON1 enzyme activity might also be changed.37 There is evidence of HDL becoming pro-inflammatory during the acute phase response, and the decreased of PON1 activity could be responsible for such a change.27,28
As discussed before, homocysteine-thiolactone may be the endogenous substrate of PON. Studies have demonstrated that homocysteine is an independent risk factor in PMOP,16,37,38 and homocysteine, a sulphurous amino acid, is created during the metabolism of methionine. Homocysteine is also linked with oxidative stress. Clinical studies have also uncovered a relation between plasma homocysteine and cardiovascular and other diseases. Our group has reported a role of slightly elevated hyperhomocysteinemia in increasing total peroxidase and reducing total antioxidants in PMOP. However, any one oxidant or antioxidant biomarker may not reveal overall oxidative stress, because oxidative stress builds up due to the imbalance between total oxidants and antioxidants in the body.16,31
Lv et al.39 found that postmenopausal women have excessive methylation of the promoter A area in the estrogen receptor alpha (ERα), and the methylation degree of these women was importantly higher than in premenopausal women. Moreover, this study also found a clear correlation between the level of homocysteine and the degree of methylation in the ERα gene. Studies suggest that homocysteine could improve the hypermethylation of the promoter A region and decrease ERα mRNA transcription, and this could be a significant mechanism for the pathogenesis of PMOP. The effect of estrogen on homocysteine/methionine metabolism might be an important way in elucidating its useful effects. The role of estrogen metabolites in protecting cardiovascular health has been confirmed. In addition, estrogens can also regulate homocysteine and PON1 levels.
On the basis of the results of various published studies, we can conclude that improved oxidative stress in PMOP patients may result in a pro-oxidation environment, which in turn decreases antioxidant PON1 activity and increases homocysteine levels. HDL-associated PON1 may be a significant factor in the pathogenesis of PMOP. Considering the individual association of PON with CVD, metabolic reactions between PON1 and the different agents that change its expression and activity might be a significant therapeutic target for future investigation. However, our understanding of human diseases changes rapidly. Recent literature clearly indicates the superiority of measuring quality over quantity.29 In accordance with that, markers like PON1 merit particular attention. Patients suffering from chronic diseases, including PMOP, that share the common etiopathogenesis of oxidative stress, will hopefully benefit from antioxidant solutions. Great effort is expended to improve synthetic products to enhance the PON1 activity of HDL.40 On the other hand, follow-up of any antioxidant therapy necessitates measurement of enzyme markers like PON1. Currently, researchers face the problem of stability of the enzyme and standardization. Further studies are required to explore more regarding PON1 enzyme and human diseases.

 XML Download
XML Download