

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Extracellular matrix (ECM) is composed largely of elastin and collagen and serves many functions essential for cardiovascular wall homeostasis,1,2 and ECM remodeling is one of the underlying mechanisms in cardiovascular disease (CVD). Cardiovascular cells and CVD-related inflammatory cells produce a large number of proteases, such as serine proteases (SPs), cysteine proteases, and matrix metalloproteinases (MMPs).3-6 Among these proteases, MMPs and SPs have been believed to contribute to the ECM remodeling in CVD development and progression processes.1,7,8 However, the inhibition of MMPs and SPs has been found to result in incomplete suppression of cardiovascular remodeling in humans and animal models,9-13 which suggests that other proteases may also contribute to the pathogenesis of CVD.
Lysosomal cysteine proteases, generally known as cathepsins (Cats), were discovered in the second half of the 20th century14 and have been characterized in various studies, as follows. First, there are 11 human Cats (Cats B, C, F, H, K, L, O, S, V, W, and X) that belong to the papain subfamily of cysteine proteases.15,16 Second, Cats have a high homology with members of the papain family.17 In mice, 19 Cats have been discovered, including several placentally expressed Cats with no human homologue.18 Third, Cats have been known to synthesize proenzymes with an N-terminal signaling peptide that targets the protein to the lumen of the endoplasmic reticulum.8,19 Fourth, Cat activity is regulated intracellularly by stefins (stefin A and B) and extracellularly by cystatins (cystatin C, or CystC) and kininogens.20 Fifth, Cats degrade almost all intracellular and extracellular proteins through their combined activities.8 Sixth, cardiovascular and CVD-related cells show different cathepsin expression patterns.8 The cysteinyl Cats are predominantly endopeptidases located intracellularly in endolysosomal vesicles.21,22 However, Cats act in the extracellular space as well as in the cytosol and nucleus.19,23 Failing cardiac and atherosclerotic vessel tissues from humans and animals overexpress the elastolytic and collagenolytic Cats S, K, B, H, and L24-27 but show no changes in levels of their endogenous inhibitor, CystC.24,28 This suggests a shift in the balance between cysteine proteases and their inhibitor that favors remodeling after cardiovascular injury. Cats may therefore be pharmacological targets in patients with cardiovascular injury.29-31 Circulating Cats or/and CystC are also potential biomarkers for detecting ischemic heart disease.32-35 This review article examines several issues concerning the biological roles and molecular functions of cysteinyl Cats in vascular pathological processes, especially with respect to their potential application as diagnostic or prognostic markers and drug targets.
CYSTEINYL CATS AND CYSTC IN CVD
The pathogenesis of CVD involves substantial proteolysis of cardiovascular extracellular proteins. Three families of proteolytic enzymes may participate in this process, including MMPs, SPs, and cysteinyl Cats. The roles of the former in various CVDs have been covered by recent comprehensive reviews.1,2,4 In this review, we consider the role of Cats in CVD in greater detail. The sections below describe the Cats involved in several atherosclerosis-based artery diseases and their complications, especially with respect to their potential application as prognostic biomarkers and drug targets to prevent CVD.
1. Cysteinyl Cats in atherosclerosis
Cats K and S were the first cathepsins found to be expressed in human atherosclerotic lesions more than a decade ago.26 Increases in the amounts and activities of Cats S and K, as well as changes in the abundance of CystC, have been shown to accompany the development and progression of atherosclerotic lesions during vascular remodeling.36 Liu et al. reported that CatS is overexpressed in the endothelial cells (ECs) lining the lumen of the arterial wall and in microvessels inside plaques.14 In a mouse model, Cats S, K, and L were increased and localized in macrophages or lipid-rich areas of diet-induced atherosclerotic lesions.28,37,38 Interestingly, deficiency of leukocyte CatS considerably altered plaque morphology, with smaller necrotic cores, reduced apoptosis, and decreased smooth muscle cell (SMC) content and collagen deposition.39 Deficiency of CatS in the whole body resulted in a 60% reduction of atherosclerotic plaque area and preservation of elastic laminal breakdown in low-density lipoprotein receptor-deficient (Ldlr-/-) mice.40 Furthermore, deficiency of CatS led to reduced SMC contents, collagen contents, and fibrous cap thickness.40 Recently, Rodgers et al. demonstrated that atherosclerotic plaque area and the number of plaque ruptures are lower by 46% and 73%, respectively, in CatS-/-/ApoE-/- mice than in control single genetic mice.41 Therefore, most of these beneficial pathologies obtained from the Cat genetic intervention mice are associated with Cat-mediated elastase and collagenase activities. This notion is further supported by direct evidence that genetic deletions of CatK and CatL reduce diet-induced atherosclerotic plaque formation and preserve vascular wall structure in established mouse models of Ldlr-/- and ApoE-/- mice.42,43 Furthermore, advanced plaques of the double knockout mice show an increase in collagen content and are less prone to rupture than are those of ApoE-/- mice.42,44
2. Cysteinyl Cats in aneurysm
The growth and rupture of abdominal aortic aneurysms (AAAs) result from increased elastin turnover, a process that critically depends on specific elastases that cleave multilayer elastic laminas.45 Cysteinyl Cats K, L, and S are among the most potent mammalian elastases,5,29,46,47 and human atherosclerosis and AAA lesions contain high levels of these proteases. In contrast, their endogenous inhibitor CystC is deficient in these lesions.48 Aortic tissue extracts of AAA patients had higher levels of Cat-dependent elastolytic and collagenolytic activities than did those of patients with aortic occlusion diseases, but CystC levels were regulated inversely.49 Deficiency of these proteases protected mice from diet-induced atherosclerosis,40 whereas CystC-deficient mice had enlarged aortic diameters.48,50 Aortic tissues from patients with growing AAAs and ruptured AAAs contained significantly higher Cat mRNA and protein levels than did control aortas,45 which suggests that cysteine proteases play essential roles in aortic wall remodeling. Recently, using gene deletions of CatK or CatL, Sun and colleagues demonstrated clearly that both Cats contributes to AAA formation by promoting lesion inflammatory cell accumulation, angiogenesis, vascular cell apoptosis, and elastin degradation and by affecting vascular cell protease expression and activities.51,52 Collectively, these findings suggest that increased Cat expression in human AAA lesions is not just secondary to the disease but also participates directly in its pathogenesis.51 Note that Bai and colleagues reported that CatK deficiency had no effect on AAA formation.53 Therefore, although cathepsins may contribute to AAA via more than one mechanism, the contradictory observations between the Bai and Sun groups could be the result of the groups using different experimental models. On the basis of the findings from both groups,51,53 Sun and colleagues raise the possibility that angiotensin-II (Ang-II) infusion enhances the numbers of peripheral CD4+ CD25+ T cells and Lg6G+ leukocytes in CatK-/-/ApoE-/- mice and increases the infiltration of CD45+ leukocytes and Mac-3+ macrophages in AAA lesions of these mice.53 The Ang-II-induced increase of inflammatory cells in CatK-/-/ApoE-/- mice in both peripheral and AAA lesions may have compensated for the effect of CatK deficiency, thereby obscuring the difference in AAA formation between CatK+/+/ApoE-/- and CatK-/-/ApoE-/- mice.52
3. Cysteinyl Cats in angiogenesis
Like the MMP family, there is growing evidence for specific intra- and extracellular functions for lysosomal Cat enzymes, which have been shown to critically influence tumor- and ischemia-related angiogenesis.6,54-56 The results of a previous study showed that high expression of Cats in endothelial progenitor cells (EPCs) was a prerequisite for their invasive capacity and facilitated the homing of EPCs to ischemic vasculature.57 Using established animal models of retinal and choroidal neovascularization, Shimada and colleagues demonstrated that both pharmacological and genetic interventions of CatL resulted in a significant decrease of intraocular neovascularization.58 EPCs from type 2 diabetes patients resulted in a profound reduction in CatL expression and its activity as compared to EPCs derived from healthy controls.59 These findings indicate that CatL expressed in EPCs plays a critical role in intraocular angiogenesis and suggest a potential therapeutic approach of targeting CatL to treat neovascular ocular peripheral artery diseases. In addition, CatK-/- has been shown to impair angiogenesis and tumor cell proliferation and angiogenic islet formation and the growth of solid tumors, whereas the absence of its endogenous inhibitor CystC-/- results in opposite phenotypes.55 Furthermore, CatS deficiency affects the production of type IV collagen-derived anti-angiogenic peptides and the generation of bioactive pro-angiogenic γ2 fragments from laminin-5, revealing a functional role for CatS in angiogenesis and neoplastic progression.55
4. Cysteinyl Cats in complications: restenosis, rupture, thrombosis, and calcification
Vascular diseases, including atherosclerosis, angioplasty-induced restenosis, vessel graft arteriosclerosis, and hypertension-related stenosis, remain the most prevalent cause of death in the developed world.60 Restenosis limits the long-term beneficial effects of percutaneous coronary intervention (PCI) and related procedures.60 Despite significant improvements in PCI technology, restenosis remains the major limitation of percutaneous revascularization techniques, with peak occurrence 1 to 3 months after successful dilation.61 Angioplasty has been shown to result in a change in luminal size and constrictive remodeling.3 As mentioned, Cats contribute to ECM degradation, which suggests a possible role for Cats in neointima formation and restenosis.3 We previously reported that the levels of Cats S and K mRNAs and proteins were increased in the carotid arteries in response a balloon injury, whereas CystC mRNA and protein showed no change.36 Immunostaining showed that increased levels of both Cats were localized in SMCs and infiltrated macrophages.36 Similarly, the neointima had higher levels of CatS mRNA and protein than that in uninjured control iliac arteries in a rabbit balloon-injury model.62 CystC mRNA and protein expression were only minimally up-regulated.62 These data indicate the importance of maintaining a fine balance between, and regulating, Cats and Cysts; disruption of this balance results in a pathological state due to deficiency or excessive degradation of collagen and other components of the cardiovascular extracellular protein.8 This notion is further supported by the results of enzyme assays showing that extracts of balloon-injured carotid arteries show an increase in elastolytic and collagenolytic activity.36 Furthermore, it has been demonstrated that CatS and CatK degrade collagen type I, fibronectin, and laminin, and that SMC transmigration through a basement membrane matrix gel can be inhibited by the selective Cat inhibitor morpholinurea leucine-homophenylalanine-vinylsulfone-phenyl (LHVS) or the broad-spectrum Cat inhibitor trans-epoxysuccinyl-L-leucylamido-(4-guanidino) butane (E64).24,26,38,46,47
It is widely believed that rupture of a vulnerable atherosclerotic plaque and related thrombosis leads to acute coronary events and stroke.63 The vulnerable plaque is generally composed of an atrophic fibrous cap, a lipid-rich necrotic core, the accumulation of inflammatory cells,64,65 and imbalance between extracellular matrix synthesis and degradation resulting in decreased extracellular matrix protein content and increased proteinases, including MMPs and SPs.37,63-66 Previously, the lack of useful animal models for this exact set of conditions limited the ability to explore the exact mechanisms of the plaque rupture. In 2006, however, our group developed a murine model of human plaque rupture that is simple, fast, and highly efficient.63 This model can help us not only to understand the mechanism of human plaque rupture but also to assess various already-known and as-yet-unknown agents in the future. Accumulating evidence shows that vascular cells (SMCs and ECs) and infiltrated macrophages and the derived foam cells act as the major cell source for the protease Cats (including CatS, CatK, CatL, CatB, and CatF) in animal and human atherosclerotic palques.5,26,37 Fibrous cap thickness has been directly associated with plaque vulnerability. Deficiency of CatS in the whole body results in a significant reduction of atherosclerotic plaque area and preservation of elastic laminal breakdown in Ldlr-/- mice.40 Furthermore, deficiency of CatS leads to reduced SMC contents, collagen contents, and fibrous cap thickness.40 CystC/ApoE double-deficient mice consistently have increased lesional SMC and collagen contents and better-developed fibrous caps than do controls.50 However, the possible involvement of cysteinyl Cats in plaque rupture requires further examination. Furthermore, to date, no direct experiment has tested for a role of these proteases in thrombosis during atherogenic complications. In addition, limited studies have shown a relationship between atherosclerotic lesion calcification and the Cat family. Previous studies have reported that macrophage-derived elastases such as elastolytic Cats S and K in collaboration with MMP-9 degrade medial elastin, which favors calcification through an increase of elastin polarity that in turn enhances elastin affinity for calcium.67,68 A recent single study has demonstrated that CatS-/- mice have provided new insights into the pathobiology of arterial calcification and have aided the investigation of novel therapeutic strategies to reduce the onset of cardiovascular events and thus mortality.69 However, further studies are needed to investigate these issues.
CYSTEINYL CATS: MECHANISMS OF ACTION ON MOLECULAR AND CELLULAR LEVELS
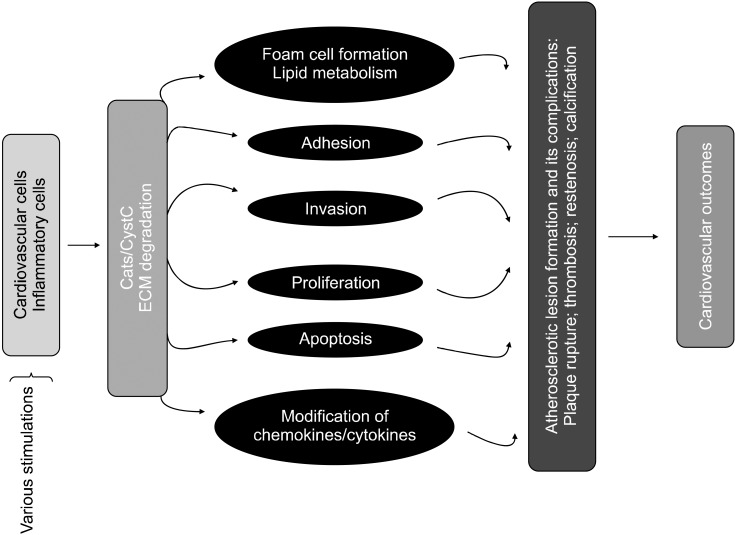
It is well known that cathepsins are implicated in CVD through their activation, liberation, and modification of angiogenic growth factors, cytokines, and proteases associated with degradation of lipid metabolism, cell events (migration, invasion, proliferation, and apoptosis), angiogenesis, and matrix protein remodeling (Fig. 1). Here, we highlight some of the mechanisms by which Cat expression and activity influence ECM metabolism, cellular functions, and inflammation on molecular and cell biological levels. By influencing these processes, cysteinyl Cats contribute to CVD, including atherosclerosis-based vascular disease and its implications.
1. Gene regulation
Cardiovascular cells and CVD-related inflammatory cells account for most of the cysteinyl Cat expression in CVD.8 CatS is expressed at low levels under serum-free conditions in cultured rat, calf, and human SMCs.46 Quantitative immunoblot analysis and polymerase chain reaction show high levels of CatS mRNA and protein in cells treated with either cytokine.26,46,70 Furthermore, increased cathepsin expression is associated with enhanced elastolytic and collagenolytic activity that is largely sensitive to a specific inhibitor of CatS (LHVS) or a nonspecific inhibitor of Cats (E64); cysteinyl Cats are therefore important in proteolytic activity in inflamed SMCs. Interferon-γ regulates CatS and CatL in macrophages.71 In cultured cardiomyocytes and macrophages, Cat expression and activity are increased by Ang-II and H2O2; these changes are moderated by apocynin, a nicotinamide adenine dinucleotide phosphate (NADPH) oxidase inhibitor.29,37 H2O2 stimulates the elevation of cystatin C protein in conditioned medium of cardiomyocytes.13 Recently, we reported that Ang-II promoted CatS expression via mineralocorticoid receptor activation in vivo and in cultured podocytes.31 These findings, together with our recent finding that none of the common inflammatory cytokines and hormones affects CatK mRNA levels in cultured cardiovascular cells and inflammatory cells, suggest that CatS/CystC, which is released from cardiomyocytes, interacts with ECM proteins, a process that is likely associated with the development of CVD in response to inflammation and oxidative stress.
2. Proteolysis
Cysteinyl Cat-mediated extracellular protein degradation contributes to a variety of physiological and pathological conditions of the cardiovascular system.8 Cats have been shown to localize on cell membranes or in endosomal/lysosomal vesicles or to be secreted into the extracellular space,19,26,38 which suggests that their enzymatic substrates and functions might change along with their localization. Recently, we demonstrated that active CatS colocalized with integrin ανβ3 on the SMC surface and played an important role in SMC-mediated matrix protein degradation.46 Accumulating evidence shows that active Cats can degrade the protein components of basement membranes and the interstitial connective matrix, including elastin, fibronectin, laminin, and many types of collagens.46,47,62 The data from gene deletion and transgenic mice studies provide direct evidence of Cat molecular function.40,54 These studies established that Cats are not simply redundant, homeostatic enzymes involved in the turnover of ECM delivered to the lysosome by endocytosis or autophagocytosis, but are critically involved in the proteolytic processing of specific substrates in CVD processes.
3. Cellular functions
It is well established that specific adhesion molecules expressed on the surface of vascular ECs, e.g., vascular cell adhesion molecule-1, intracellular adhesion molecular-1, and chemoattractant molecules, such as macrophage chemoattractant protein-1, play a critical role in leukocyte recruitment from the circulation by adhesion to the endothelium as the first step of inflammatory diseases such as atherosclerosis.72 Until now, there has been no direct evidence that cysteine Cats play any role in regulating these adhesion molecules or in leukocyte adhesion. The authors of one previous study reported that cathepsin S deficiency reduces the serum levels of these molecules of mice with diet-induced atherosclerosis.40 Therefore, CatS may act like MMPs and release adhesion molecules from the surface of ECs.
Following adhesion transmigration through the endothelial layer and basement membrane, monocytes become macrophages, proliferate, and become lipid-laden foam cells.72 Type IV collagen, laminin, and fibronectin are major components of the vessel subendothelial basement membrane. Macrophages derived from animal and human monocytes have been shown to express and secrete substantial amounts of active CatS, CatL, and CatK, which can degrade these subendothelial basement membrane components.72 On the other hand, under normal conditions, vascular SMCs in the tunica media of blood vessels are quiescent and are embedded in a network of elastin-rich ECM that acts as a barrier to SMC migration and proliferation.36,73 Early in the formation of the thickened intima, as in atherosclerotic and neointimal lesions, SMCs that migrate from the tunica media into the developing intima must penetrate the internal elastic lamina.36 Destruction of the aortic media and supporting lamina through the degradation of elastin is also an important mechanism in the formation and expansion of aortic aneurysms.74 SMCs in the arterial wall are believed to be involved in this vascular remodeling through the production of various proteases, and degradation of the elastin component is believed to be the result of a proteolytic cascade that involves the cooperation of SPs, MMPs, and cysteinyl Cats.11,12,36,75,76 Recent studies have demonstrated that gene disruptions of CatS or CatK prevent the degradation of elastic lamina in aortic atherosclerotic lesions.40,42 Moreover, CatS- or CatK-null SMCs yielded similar results,40,42 which suggests that these Cats may participate in elastin-rich ECM degradation and SMC migration during the development and growth of neointima-related stenosis and atherosclerotic plaque.
CYSTEINYL CATS IN THE TREATMENT AND BIOMARKERS OF CVD
1. Treatments
Over the past decade, several pharmaceutical companies have become interested in Cat inhibitor development. Nevertheless, there has been no report on the therapeutic value of Cats in CVD. A single previous laboratory study showed that CatK inhibition reduced body weight and improved glucose metabolism in mice.77 E64d (l-3-trans-carboxyrane2) is a broad-spectrum inhibitor of cysteine proteases that inhibits the activity of several Cats (including CatS, CatK, CatB, and CatL).54 To our knowledge, E64d was applied for the first time to evaluate Cat inhibitor-mediated vasculoprotective effects on cardiac and renal injuries in response to salt-induced hypertension. E64d has been shown to reduce the extent of both cardiac and renal fibrosis with the decreased elastolytic activity in heart failure rats.29,31 E64d also suppressed the degradation of the intramyocardial coronary elastin lamina in heart failure rats. However, E64d had no significant effects on MMP-2 or MMP-9 expression or activation. These findings, coupled with previous findings that Cats are secreted into the extracellular space, showing potent collagenolytic and elastolytic activities, indicate that E64d prevents cardiovasculorenal fibrosis and remodeling through a mechanism that could be associated with the reduction of Cat-dependent ECM degradation. Recently, the Samokhin group reported that ApoE-/- mice treated with CatS inhibitor displayed fewer elastic lamina breaks, infiltrated macrophages, and buried fibrous caps and showed smaller atherosclerotic plaques.78 However, limited information is available regarding these cysteine protease inhibitors in treating cardiovascular diseases.
There are advantages to employing drugs that target Cats as part of the proteolytic pathway.8 Ang-II inhibition has been shown to decrease CatS and CatK as well as MMP-2 and MMP-9 expression and to improve advanced atherosclerotic lesion formation and atherosclerotic plaque instability.79 We reported that Ang-II antagonism suppressed the expression of the CatS and CatK proteins and helped to improve cardiac remodeling and dysfunction in a salt-induced hypertensive rat model.29,80 Recent studies demonstrated that statins prevent diet-induced cardiac and renal damage via a reduction in oxidative stress production mediated by the Ang II signaling pathway and in CatS expression or activity in animal models.31 Although only limited basic findings are available, the results that are available favor the notion that the cysteinyl Cats might be the best targets of drugs to prevent CVD in clinical trials. To our surprise, a few studies have reported that CatL-null mice exhibit a human cardiomyopathy-like phenotype.81,82 Therefore, several questions remain. These include how CatL deficiency and the observed alteration of the acidic organelle change intracellular signaling toward induction of a hypertrophic response with subsequent dilation of the heart. This has prompted basic and clinical scientists to investigate whether genetic or pharmacological interventions to Cats can produce beneficial cardiovascular actions in response to various injuries.
2. Biomarkers
Recent studies highlight the evaluation of serum Cat levels as a diagnostic tool for CVD, much like the use of MMP. Among Cat family members, CatS and CystC are most often studied as possible tools for treating various diseases.32,35,83 Liu et al. were the first to report an increase in serum CatS in patients with ischemic heart disease.32 More recently, two studies showed that CatL can also be used as an independent biomarker in ischemic heart disease.33,84 CystC has been recognized as a sensitive marker for potential renal dysfunction and injury and as an independent predictor of cardiac outcomes in patients with heart failure.85,86 High serum CystC levels are associated with increased left ventricular hypertrophy and dysfunction.87 A comparison of the serum levels of patients with AAA to those of patients with normal aortas showed decreased levels of serum CystC in the patients with AAA.48 These data suggest that measurement of serum Cats or CystC levels may be helpful in the diagnosis of cardiovasculorenal disease, but this requires further exploration.
CONCLUSION
Many data from clinical and basic studies of atherosclerosis-based CVD support a role for Cats in these diseases. Pharmacological inhibition of Cat is now being investigated in human trials for CVD, such as for AAA. Because the prevalence of atherosclerosis-based CVD and its complications is increasing and may coincide with the growth of the aged population, dual therapy targeting these diseases may be considered as a future therapeutic strategy. Until now, however, no data have been available on the effect of these inhibitors in CVD. New research will determine whether selective and reversible Cat inhibitors will be pharmacologically effective and physiologically safe in treating human CVD. The current quest for Cats as a biomarker therefore seems a reasonable goal in CVD research.
 XML Download
XML Download